
单细胞测序可用于测量细胞的多种特征模态(multi-modality),包括基因表达(mRNA/gene expression),染色质可访问性(chromatin accessibility),以及蛋白质含量(protein abundance)[1]。对于同一生物组织的不同组单细胞测序实验,其结果往往会受到不同实验环境以及操作的影响,进而呈现出不同的表达特征[2]。另一方面,不同的实验往往也会有不同的侧重点,进而测量不同的生物特征。单细胞数据融合方法其目的在于融合不同实验对同一生物组织的多模态测量,消除不同实验间因环境差异造成的测量偏差。目前的单细胞数据融合方法往往只能处理特定的数据结构,其泛用性受到较大影响。该研究设计了用于单细胞数据融合的新型矩阵分解算法。该算法进一步拓宽了单细胞数据融合的适用场景,同时该方法可以在融合数据的同时从不同模态提取出标定细胞种类的关键特征信息。张秀苇团队将该算法应用于多种已知单细胞数据,并成功融合了多种多特征人体外周血单个核细胞数据,小鼠大脑皮质数据,人体骨髓数据,以及人体脾脏数据。同时张秀苇团队运用计算生成的模拟数据系统性分析了该方法在不同融合场景下的表现效果。实现结果表明了该方法相比于其他方法在应用场景和融合精度上的优越性。

scMoMaT将单个单细胞数据测量矩阵分解成细胞成分矩阵,特征成分矩阵(mRNA, protein, chromatin accessibility),以及关联矩阵。其中细胞成分矩阵可以被2D可视化并用于分离不同细胞种类,特征成分矩阵可用于提取不同种类细胞的不同种特征成分,最终完成细胞种类的标定。对于多个细胞数据测量矩阵,scMoMaT可挖掘出不同矩阵之间细胞或者特征的关联信息,从而完成对于实验误差的校准。
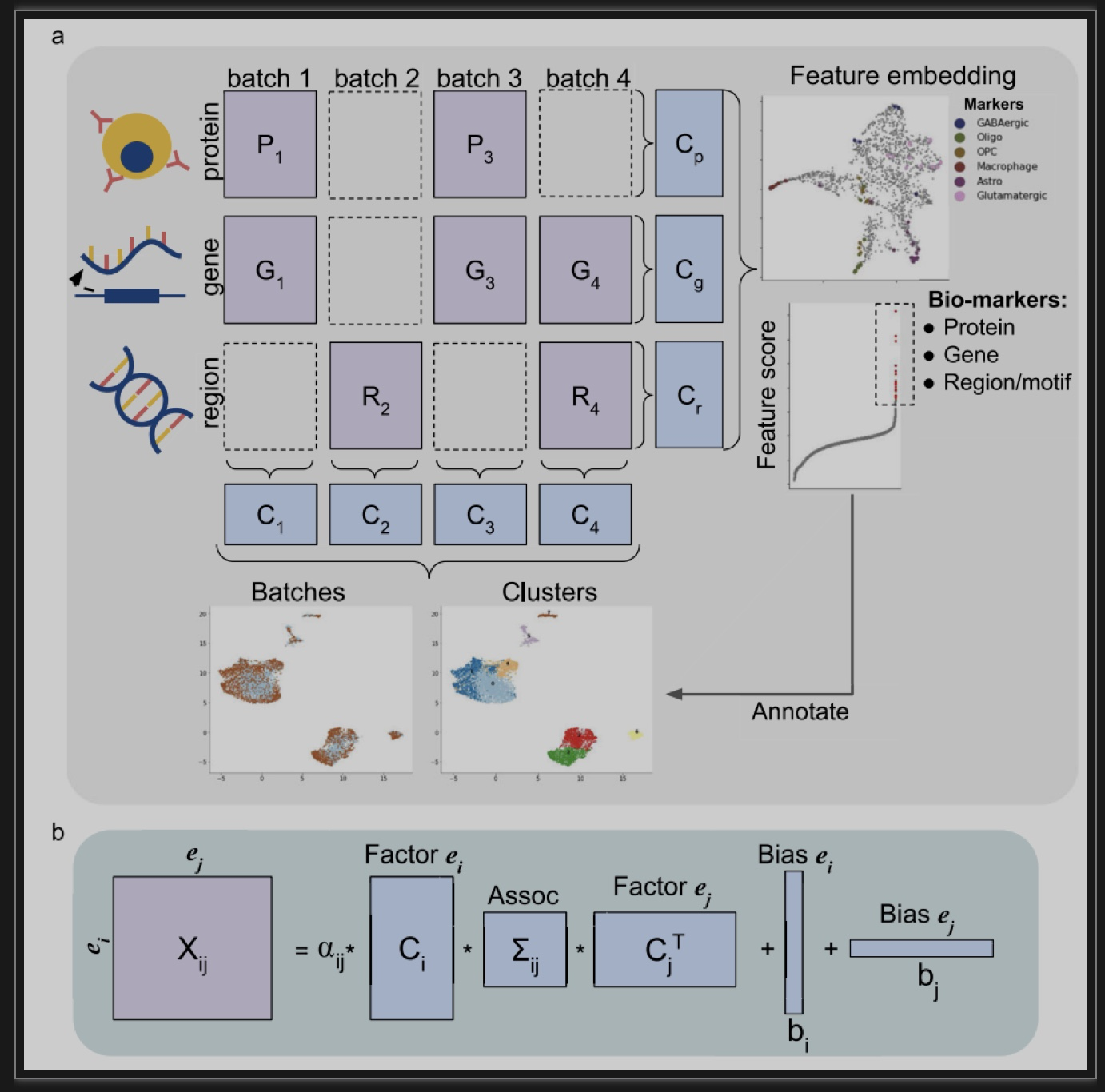
图1. scMoMaT的系统框图。a. 对于输入的多个单细胞测量矩阵,scMoMaT可分离出细胞组成信息以及特征信息。b. 对于每个个单细胞矩阵,scMoMaT都将其分解成不同的成分矩阵。
(图源:Zhang, Z. et al., Nat. Commun., 2023)
张秀苇团队将该方法应用于人体外周血单个核细胞数据集,该数据组包含3种模态信息 (mRNA/gene, protein, chromatin accessibility/region),以及4组实验测量(图2a)。scMoMaT分离出的细胞成分矩阵被用于可视化展示以及细胞种类分离(图2b,c,f),同时scMoMaT提取的特征信息被用于标定细胞种类(图2e)。与其他方法的横向比较展示出scMoMaT在细胞成分分离以及实验误差校准方面达到或优于现有的最佳方法(图2d)。

图2. scMoMaT在人体外周血单个细胞数据组上的表现结果。a.数据组的数据构成。b, c, e, f. 细胞成分矩阵的可视化结果。d.与其他方法横向比较的精度分数展示。g, h, i. scMoMaT提取出的特征成分信息成功反映了真实细胞组成。
(图源:Zhang, Z. et al., Nat. Commun., 2023)
该研究拓展了已知单细胞数据融合算法的适用场景,提升了已知算法的融合精度,并实现了特征提取与细胞数据融合的同步进行。在多种已知单细胞数据集和模拟数据集上的测试验证了方法的优越性。同时该方法也初步探索了在数据融合的过程中学习不同种特征的关联,进一步的研究将着重于更精确的提取出不同种特征的关联网络,以便于进一步分析不同细胞组成的内在调节机制。
