
据世卫组织统计,2020年原发性肝癌是全球范围内发病率排名第6位的恶性肿瘤,每年新发病例数达90.6万人,死亡83.0万人。肝细胞癌(Hepatocellular carcinoma,HCC)占所有原发性肝癌的85%~90%,是全球癌症死亡的主要原因之一,而且恶性程度高,现有一线治疗手段索拉菲尼,乐伐替尼,以及贝伐珠单抗和阿替利珠单抗联合疗法[1],仍不能满足长久的生存获益,存在巨大的未满足临床需求。研究表明,FGF19-FGFR4信号通路的异常激活在HCC的肿瘤发生过程中发挥重要功能[2],因此,FGFR4抑制剂也成为HCC新的潜在治疗靶点。包括和誉医药独立自主研发的Irpagratinib (ABSK011)在内,全球已有多款FGFR4高选择性抑制剂,如Fisogatinib(BLU-554)处于临床开发阶段,并在携带FGF19过表达的HCC患者中显示出初步的临床疗效[3],然而,总体临床反应率和反应持续时间仍比较有限,可能由于发生获得性耐药而导致,这也对单药治疗提出了挑战。因此,深入认识并阐述FGFR4的耐药机制并寻找更强效的治疗方案,对FGF19过表达的HCC患者的临床获益十分重要。

该研究通过体外长时间培养Huh7肿瘤细胞系并暴露于高浓度FGFR4抑制剂BLU-554药物中,成功建立了针对BLU-554耐药的细胞模型(Huh7-A1, Huh7-A2, Huh7-A3),并发现耐药细胞对于其他FGFR4抑制剂包括FGF-401, H3B-6527和Erdafitinib同样耐药。随后,作者利用phospho-RTK阵列实验发现EGFR磷酸化水平在所有3个耐药细胞系中上调,western blot结果也验证了三种耐药细胞株中EGFR和下游信号通路中的ERK1/2和AKT磷酸化水平均显著增加。此外,转录组测序和生物信息学分析结果进一步证明了EGFR通路激活相关signature在耐药细胞株中显著富集,进一步证实了EGFR/MAPK/AKT信号通路的激活是介导FGFR4抑制剂获得性耐药的主要机制。在明确耐药机制之后,作者在获得性耐药的细胞模型上验证了BLU-554与EGFR抑制剂吉非替尼或者厄洛替尼联用在抑制肿瘤细胞生长、促进细胞凋亡以及细胞周期阻滞上均表现出协同效应。除了与EGFR抑制剂联用,作者还进一步探索了与EGFR信号通路下游的关键蛋白SHP2抑制剂的联用,同样也观察到了相似的效果。从信号通路的分析来看,BLU-554与EGFR抑制剂或者SHP2抑制剂的联用进一步阻断了下游AKT,MAPK信号通路,从机制上解释了协同效应的发生。
除了在获得性耐药细胞模型中的发现之外,作者还通过在HCC亲本肿瘤细胞中外源添加EGF,构建原发性EGFR信号通路激活的HCC细胞模型。结果发现在多个HCC肿瘤细胞中,原发性EGFR信号通路的激活也造成了BLU-554的耐药,并且这种耐药能够被EGFR和SHP2抑制剂逆转。对下游信号通路的检测结果也证明EGF的加入激活了EGFR/AKT/MAPK信号通路,EGFR和SHP2抑制剂的加入进一步阻断了该信号通路。
此外,作者进一步进行了体内药效实验,构建了耐药细胞Huh7-A3和亲本细胞Huh7的体内异种移植肿瘤模型,在体内验证了Huh7-A3对于BLU-554的耐药性。在两个模型中均观察到BLU-554与吉非替尼联合用药组的肿瘤缩小,药效显著优于各自单药组和溶媒组。对肿瘤组织的免疫组化染色结果也表明,在联合用药组,反映肿瘤细胞增殖活性的Ki67水平显著下调,凋亡细胞明显增加。Western blot分析发现在耐药肿瘤中,联合用药组的肿瘤对ERK1/2磷酸化水平的抑制更加彻底,这与药效结果一致。
鉴于以上发现,作者进一步分析TCGA数据库中HCC患者的基因组数据,发现在FGF19高表达的HCC患者中,确实有较多的患者存在EGFR信号通路激活。这也可能是目前BLU-554等选择性抑制剂单药临床治疗FGF19高表达HCC患者时疗效欠佳的潜在原因。
代偿性旁路激活是导致酪氨酸激酶抑制剂耐药的主要原因之一[4]。2021年,仁济医院研究人员也曾在《Nature》上发表相关论文,报道了EGFR对lenvatinib耐药性的贡献,并证明lenvatinib主要通过抑制FGFR1-3信号传导与EGFR抑制剂产生协同作用。在此项研究中,作者第一次揭示了在肝癌中EGFR激活对于FGFR4耐药的贡献,验证FGFR与EGFR通路的高度相关性和交互耐药机制(图1),进一步揭示了EGFR和FGFR4抑制剂联合治疗的巨大潜力,为FGFR4抑制剂的临床开发指明新的方向。
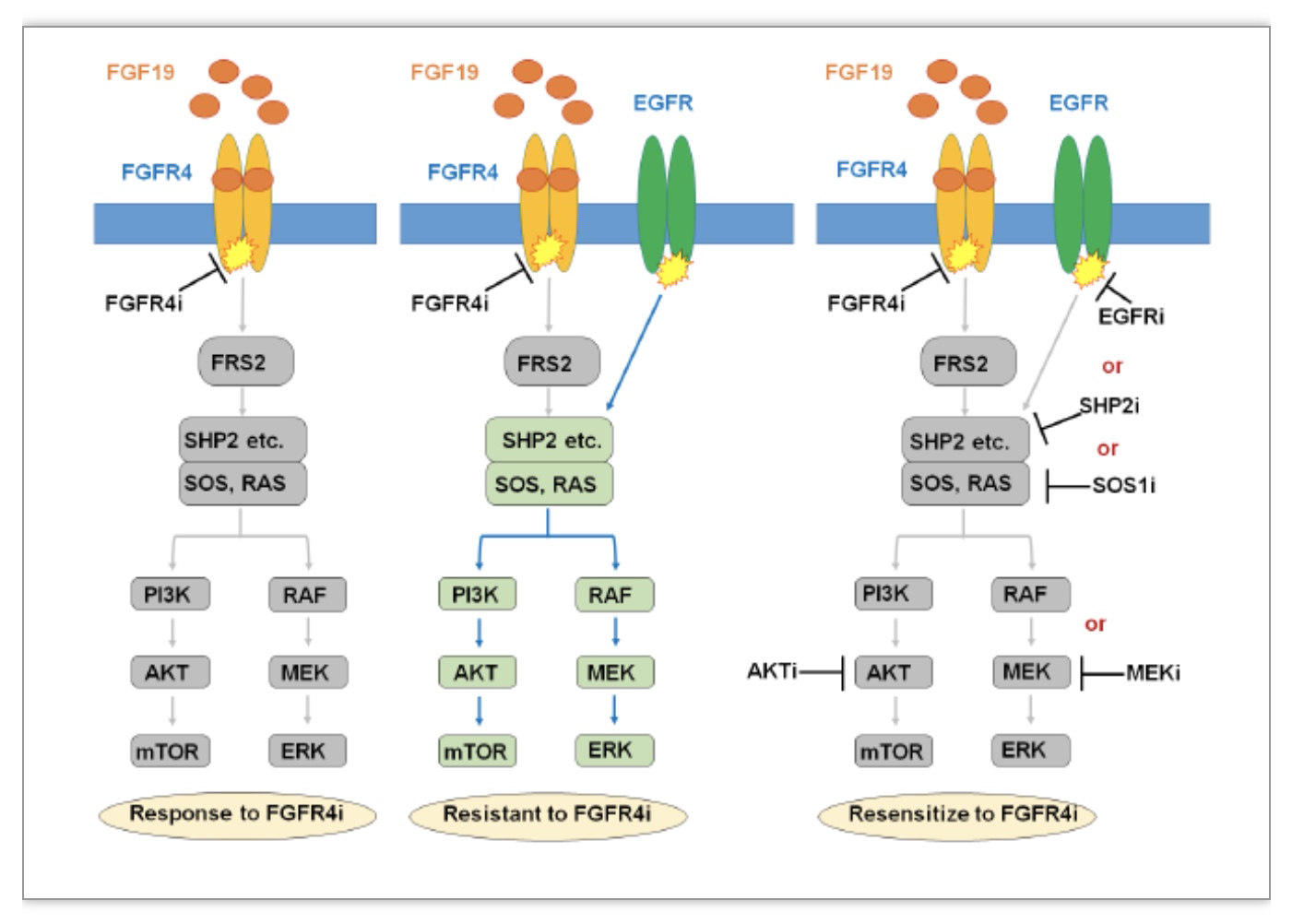
图1 EGFR激活参与FGFR4抑制剂BLU-554的耐药性产生机制
