
胚胎干细胞(Embryonic stem cells, ESCs)是一类体外培养可无限增殖、自我更新并分化为所有体细胞的细胞,也是哺乳动物早期胚胎发育研究的重要模型,被广泛用于药物筛选、细胞治疗和再生医学[1]。近年的研究表明,ESCs细胞特性的维持不但包括了胞外信号分子和胞内转录因子串联执行的信号传导、表观遗传层次的修饰,同时也需要细胞内代谢模式转化的协同参与[2-4]。糖酵解既是ESCs无限增殖的能量来源,也参与干性维持。随着ESCs分化,其糖酵解能力下降,氧化磷酸化水平增强[5, 6]。酵解代谢酶大多受维持ESCs多能性的转录因子(如Sox2, Oct4)的调控,同时酵解代谢产物又可选择性地驱动信号传导,调控干细胞的分化状态[7, 8]。LIF-Stat3信号通路在小鼠(m)ESC多能性维持过程至关重要[9],明确LIF-Stat3信号在ESC糖酵解中的潜在作用机制,将为ESC多能性维持和谱系规范调控提供更深入的见解,并对临床应用产生一定影响。建立了LIF-Stat3信号与糖酵解酶(血小板亚型磷酸果糖激酶,Pfkp)之间的连接,揭示了Pfkp调控mESC胚系分化过程的新功能。

ESCs中细胞能量代谢与细胞命运之间密切关联。确定ESCs能量来源的糖酵解与维持细胞干性的LIF-Stat3信号通路之间的内在关系,有助于干细胞特性的研究理论,促进了干细胞的临床应用。为了确定LIF-Stat3信号与糖酵解之间的交叉点,作者用shRNA沉默Stat3的表达,酵解各限速酶的蛋白水平检测结果表明Pfkp是Stat3转录抑制的潜在靶标(图1A)。随后,敲低各限速酶检测它们对ESCs多能性的影响,意外发现,作为酵解第二限速酶的Pfkp强烈抑制干性因子(尤其的Sox2)的表达(图1B),也就是说Pfkp抑制细胞干性。这似乎与“胚胎干细胞中糖酵解能力强”的观念相矛盾!对磷酸果糖激酶三亚型(Pfkl, Pfkm, Pfkp)的糖酵解能力分析结果表明,只有Pfkl的减少会使干细胞内ECAR和乙酰辅酶A的水平下降(图1C, D)。蛋白定量结果显示,在mESC中Pfkl占三亚型总量近七成,是其糖酵解通量的主要调控者(图1E)。由此推测受Stat3转录负调控的Pfkp可能参与调控干细胞分化。
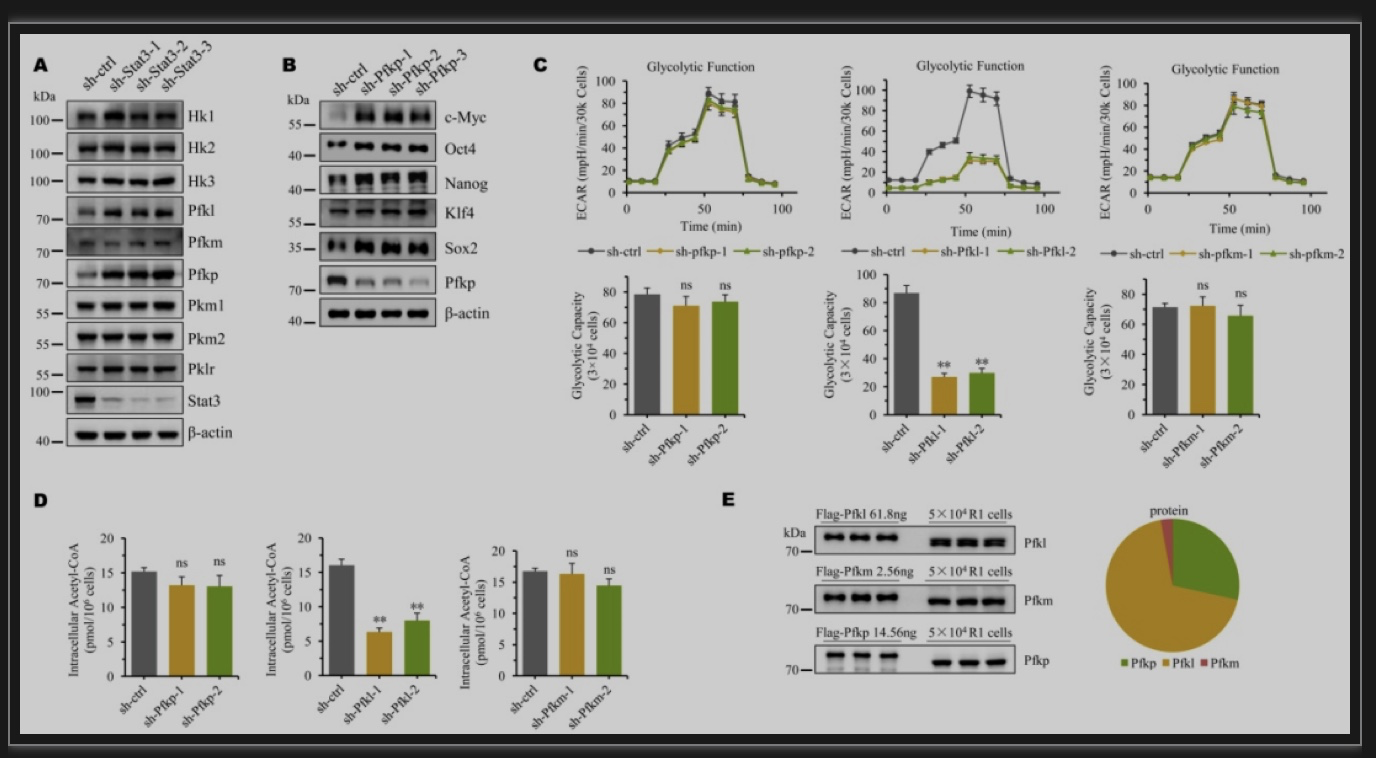
图1 Pfkp调控mESCs干性
(图源:Cao LX, et al., EMBO Reports, 2023)
为了验证Pfkp对干细胞分化的驱动作用,作者在mESC中过表达Pfkp,发现不仅核心多能性标志物的表达量整体下降,mESC还出现了与分化一致的形态变化(图2A, B)。在胚状体(EB)形成期间和LIF缺失培养过程中,Pfkp的蛋白量均上调(图2C, D)。敲减Pfkp并EB分化后的mESC转录组中存在许多参与发育和分化相关进程的差异基因(图2E)。进一步分析发现,Pfkp的减少导致许多外胚层规范基因的表达量增加,相反地,中胚层和内胚层规范基因的表达减少(图2F-H)。在定向内胚层分化的mESC细胞群中,Pfkp的减少导致内胚层规范基因(Sox1和Foxa2)阳性细胞群减少;而在神经元分化进程中沉默Pfkp会产生更多的b-III Tubulin和neurofilament-L阳性细胞(图2I-L)。这些数据共同表明,Pfkp促进mESC向内胚层分化,抑制向外胚层分化。
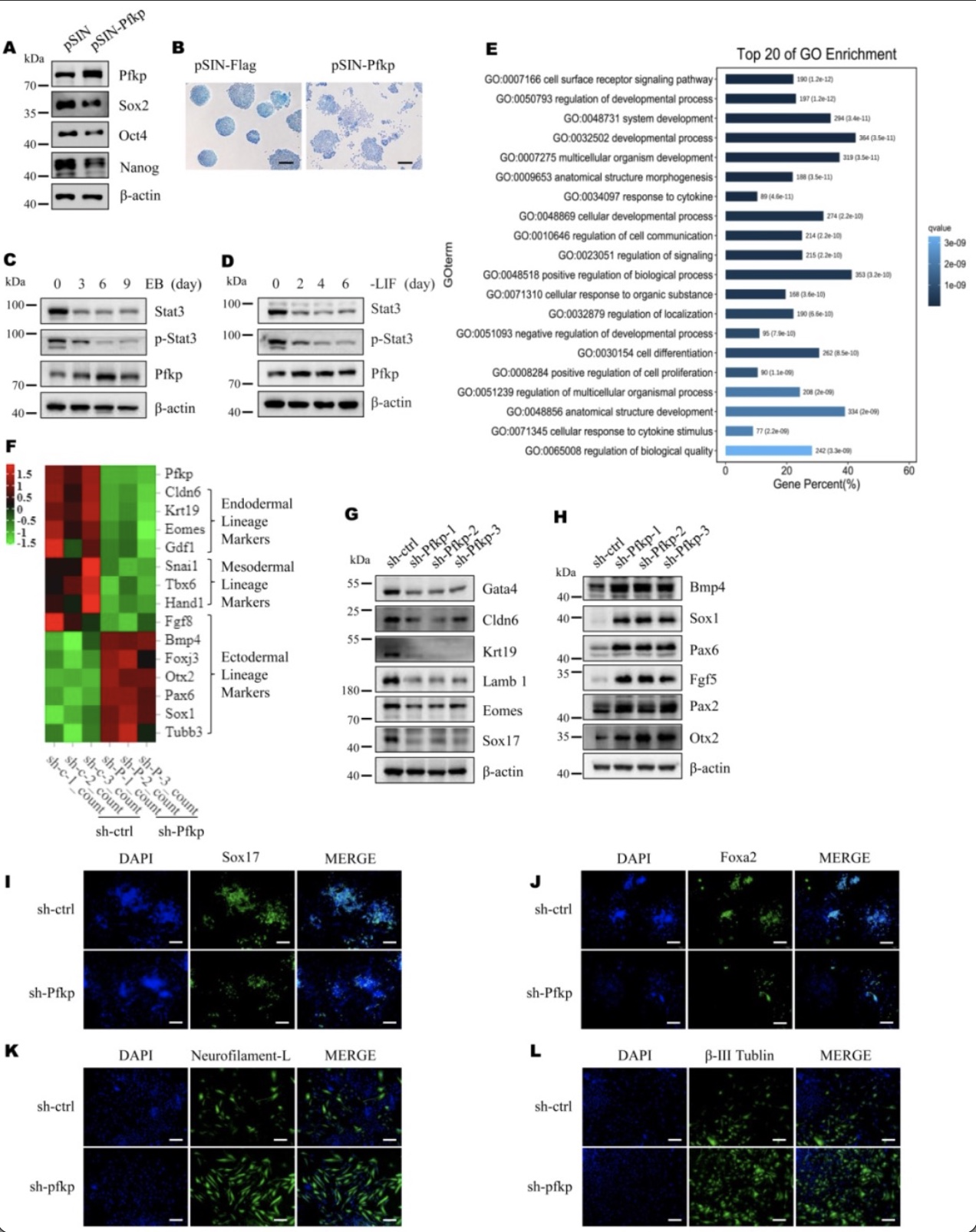
图2 Pfkp调控mESCs分化
(图源:Cao LX, et al., EMBO Reports, 2023)
鉴于Pfkp对胚层规范的广泛影响,作者推测Pfkp肯定有一个能力强大的“兄弟”。免疫共沉淀和质谱分析找到了Pfkp的互作蛋白Lin41(图3A, B)。沉默Pfkp后,Lin41蛋白量减少而其mRNA不变,而加入蛋白酶体抑制剂MG132或乳胞素(Lactacyctin)可恢复Lin41的蛋白水平(图3C, D)。过表达Pfkp后,Lin41的蛋白稳定性增强(图3E)。由此推测,Pfkp可保护Lin41免于降解。早期研究表明,磷酸化修饰可以影响Trim家族蛋白(如Trim24)的活性和稳定性[10]。作者猜想是否作为磷酸果糖激酶的Pfkp也可将其激酶活性延申到蛋白上。果然,易位表达Pfkp后,Lin41的磷酸化水平增加;而敲减Pfkp后,其磷酸化水平下降(图3F)。体外激酶实验进一步证实了Pfkp可以磷酸化Lin41(图3G)。此外,突变Pfkp的活性位点(Gly-33,ATP结合位点)后,将检测不到磷酸化的Lin41,Pfkp失去了其蛋白激酶功能(图3H, I)。为了探究是否Lin41的磷酸化与其蛋白稳定性相关联,作者用质谱鉴定出了mESC中Lin41的潜在磷酸化位点Ser-174和Ser-800(图3J)。过表达Pfkp可以降低野生型和S800A(Ser-800位点突变体)Lin41的泛素化水平,但并不影响S174A(Ser-174位点突变体)Lin41的泛素化水平。相似地,敲减Pfkp后,野生型和S800A突变体 Lin41的泛素化水平都增加,S174A突变体的泛素化水平没有明显变化(图3K, L)。这些数据表明,Pfkp磷酸化Lin41并抑制其泛素化和蛋白酶体降解。
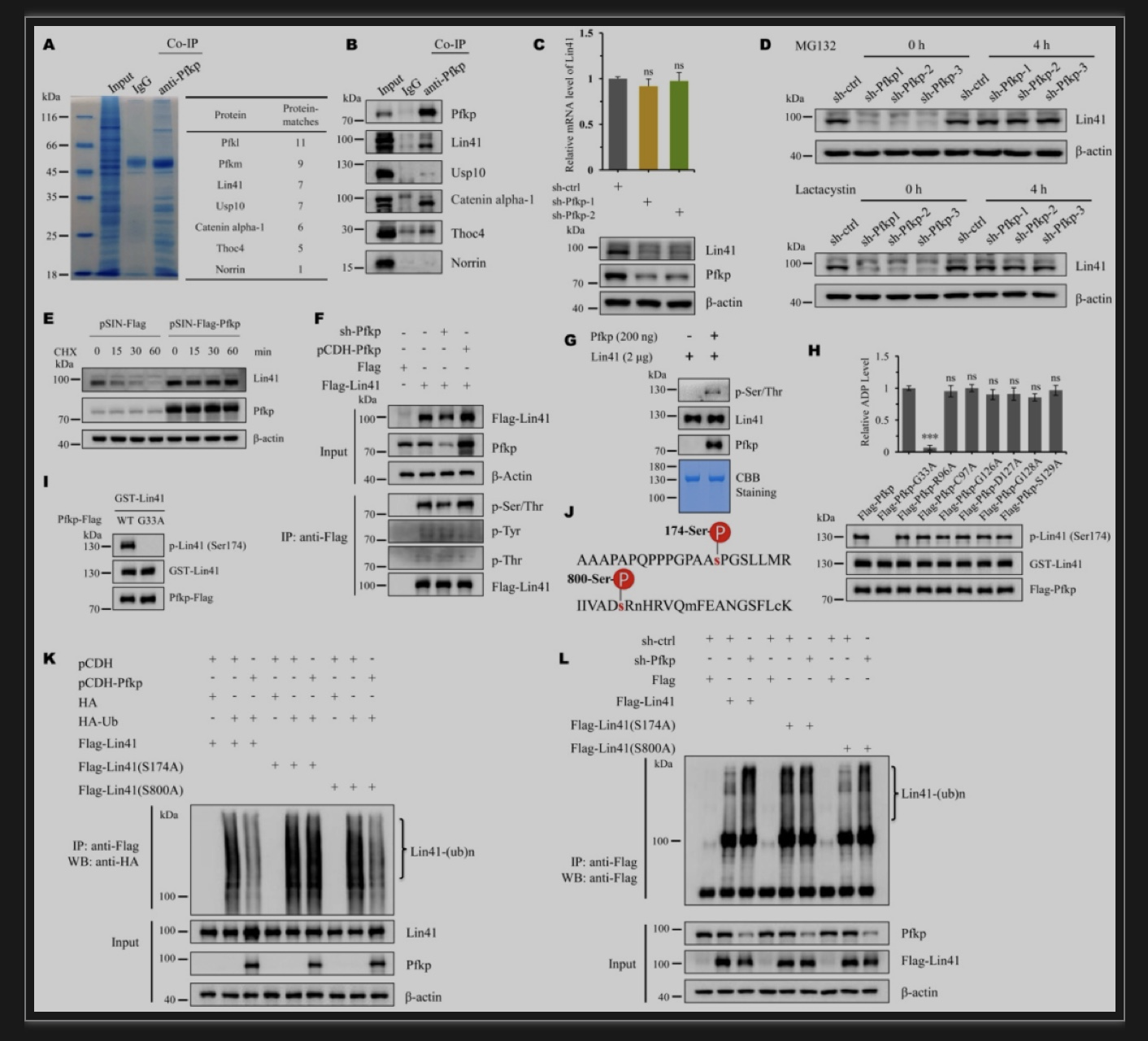
图3 Pfkp磷酸化并稳定Lin41
(图源:Cao LX, et al., EMBO Reports, 2023)
为了明确Pfkp是否是通过调控Lin41来影响胚层分化的,作者对Lin41敲减并EB分化后的mESC细胞进行转录组分析,结果表明,在前20个基因功能集中也有许多与发育和分化相关的生物进程(图4A)。Lin41的沉默同样上调了一组外胚层规范基因,抑制了许多中内胚层规范基因的表达(图4B-D)。鉴于Lin41具有结合并负调控mRNA表达的功能,作者假定Lin41和外胚层基因mRNA之间选择性相互作用。RIP和RNA-pulldown结果表明Lin41与外胚层规范基因Fgf5、Otx2、Sox1和Pax2的mRNA结合(图4E, F)。沉默Lin41和Pfkp后,此四个外胚层规范基因mRNA的稳定均增加,而这些增加又可被Lin41易位表达逆转(图4G, H)。这些结果表明,Lin41通过与外胚层基因mRNA相互作用辅助Pfkp调节mESC分化。
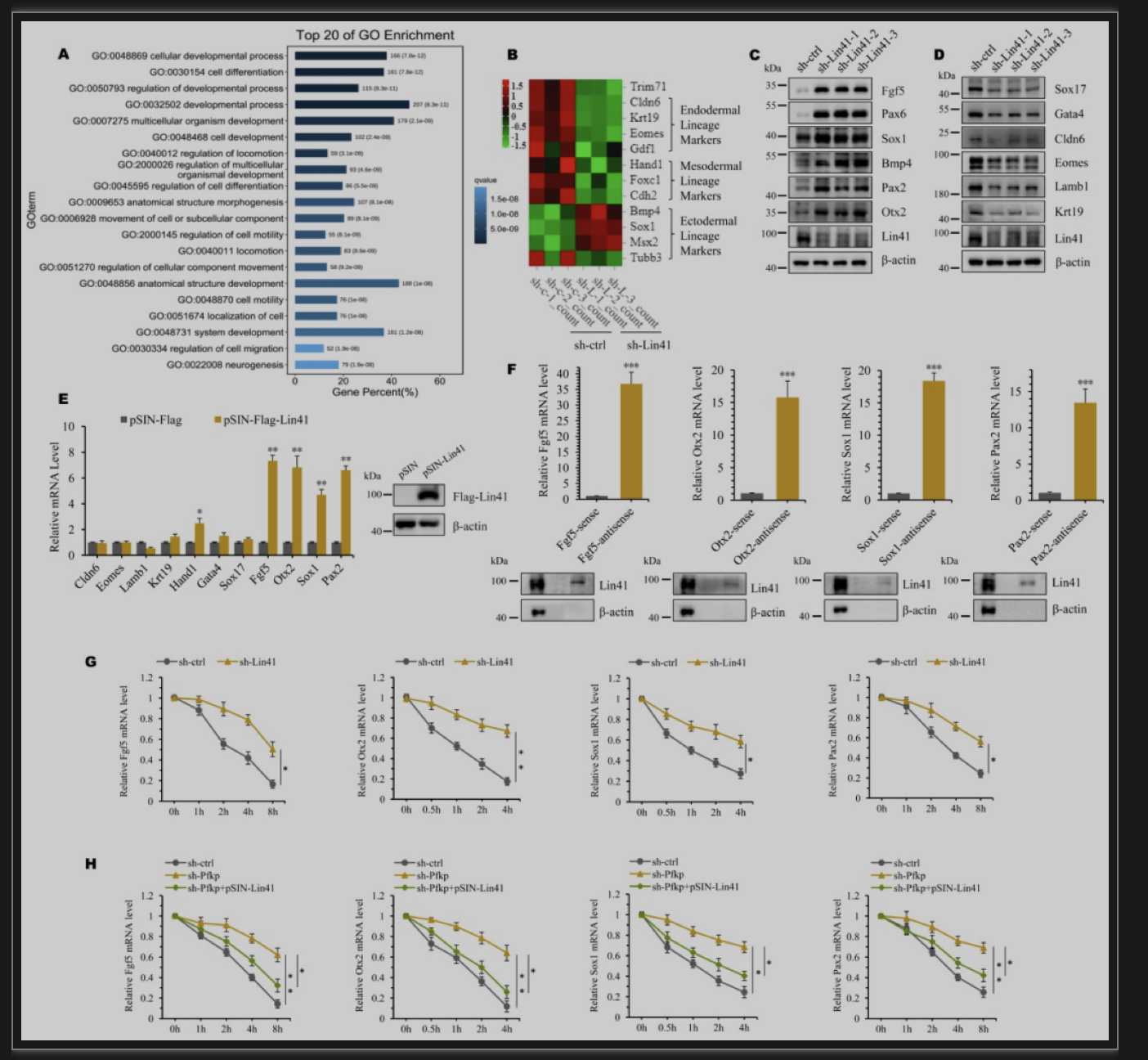
图4 Lin41结合外胚层基因mRNA
(图源:Cao LX, et al., EMBO Reports, 2023)

图5 Pfkp调控mESC分化的模式图
(图源:Cao LX, et al., EMBO Reports, 2023)
总的来说,本研究揭示了LIF-Stat3-Pfkp-Lin41通路在早期胚胎发育过程中的作用,为mESC 多能性维持的新型调节轴提供了证据(图5)。LIF介导的Stat3激活会抑制Pfkp转录,进而通过自身泛素化机制抑制Lin41的表达。解除Stat3对Pfkp的抑制,将允许Pfkp磷酸化Lin41,进而抑制其泛素化和蛋白酶体降解。稳定的Lin41进一步结合并降解参与外胚层规范的关键基因mRNA,从而利于决定内胚层谱系的mRNA的表达。因此,我们的结果为平衡谱系之间规范的分层控制机制提供了证据。但值得注意的是,关于原始多能性状态下(即非Stat3激活状态下),mESC是如何拮抗Pfkp来维持干细胞的自我更新,仍需要进一步研究。此外,Pfkp抑制核心干性因子的具体分子机制,及其与Pfkp激酶功能是否相关?也有待进一步探究。
