
肝细胞癌(HCC)是最常见的癌症之一,也是造成全球癌症相关死亡的主要原因。由病毒感染、酒精滥用或脂质积累引起的肝脏炎症在肝癌发生中起关键作用。然而,这种恶性转化的潜在机制仍不大清楚。因此,对肝癌发生过程中的炎症进行调控具有重要的科学意义和临床价值。
之前的研究发现,视黄酸诱导基因I(RIG-I)是宿主识别RNA病毒感染并诱导固有免疫细胞产生I型干扰素的关键传感器之一。RIG-I不仅在免疫细胞中表达,还在肝实质细胞中表达,表明它在肝脏功能和疾病中发挥潜在作用。不过,RIG-I在肝脏代谢、脂肪变性和癌变中具体发挥什么作用,现在还知之甚少。表明RIG-I可防止坏死性炎症及非酒精性脂肪性肝炎诱导的肝癌发生,为肝细胞癌的预防提供了潜在靶点。

研究材料与方法
技术路线
研究结果
1、肝癌祖细胞中RIG-I的表达下降驱动了肝癌发生
为了分析RIG-I减少在肝癌发生中的作用,研究者构建了肝细胞特异性RIG-I敲除小鼠(Rig-Ihep−/−小鼠,hep即hepatocyte),发现肝脏RIG-I缺失显著促进了DEN诱导的肝癌发生。DEN+CCl4诱导的肝癌发生模型也证明了这一点。这些结果表明,HcPC中RIG-I的表达下降可能参与了肝癌发生。以Rig-Ihep−/−小鼠作为对象,研究者发现,肝脏RIG-I缺失能够增强致癌的IL-6-STAT3信号通路,促进HcPC发展成为肝细胞癌。
IL-6是主要通过影响JAK2/STAT3信号通路而驱动肝癌发生的关键促炎细胞因子。研究者发现,RIG-I的N端CARD结构域能够与JAK2的JH2结构域竞争,从而抑制JAK2对STAT3分子的磷酸化。考虑到IL-6在早期诱导肝脏RIG-1表达,这些数据都说明了IL-6诱导了RIG-I与STAT3的结合。
至于背后的机制,研究者决定从翻译后修饰着手开展分析。RIG-I的K18和K146位点发生组成型的单甲基化修饰,研究者发现IL-6能够诱导RIG-I的K18和K146位发生去甲基化,而这种去甲基化能够增强RIG-I与STAT3的结合,并抑制IL-6-STAT3信号传导(图1)。通过免疫沉淀和质谱分析,研究者认为去甲基化酶JMJD4可能参与了IL-6诱导的RIG-I去甲基化。在敲除JMJD4后,RIG-I的K18和K146位没有发生去甲基化。
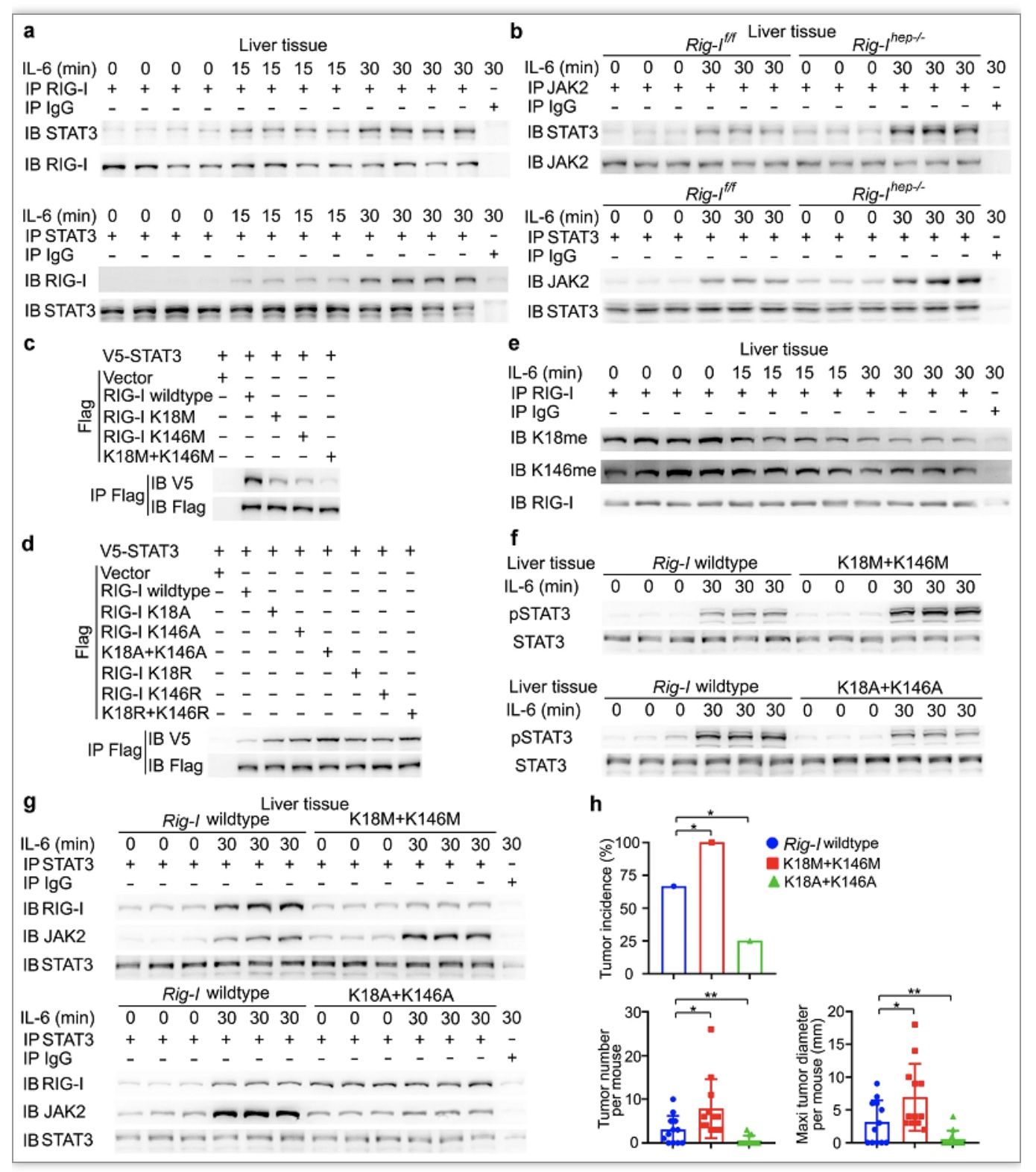
图1 IL-6诱导RIG-I去甲基化以增强RIG-I与STAT3的结合[1]
2、RIG-I在NASH诱导的肝细胞癌中起肿瘤促进作用

图2 肝细胞特异性的RIG-I缺失消除了脂肪变性和肝癌发生[1]
NASH诱导的肝细胞癌会经历脂肪变性、NASH、纤维化、肝硬化和肝细胞癌等阶段,于是研究者分析了肝脏RIG-I缺失是否会对第一步脂肪变性形成抑制。在利用HFD诱导脂肪变性后,研究者发现与对照小鼠相比,肝脏RIG-I缺失抑制了HFD诱导的脂肪变性,并显著降低了肝脏内胆固醇的堆积。同时,RIG-I敲除还能降低血清胆固醇的水平,并抑制小鼠动脉粥样硬化的进展。这些结果表明,RIG-I缺失消除了肝脏胆固醇积累,抑制了肝脏脂肪变性和高血清胆固醇诱导的疾病。
为了阐明背后的分子机制,研究人员对Rig-If/f和Rig-Ihep−/−小鼠的肝脏开展了转录组以及蛋白质分析。研究者发现,胆固醇合成的限速酶HMGCR的磷酸化在Rig-Ihep−/−肝脏中显著增加,而其磷酸化会导致失活。因此,研究者推测HMGCR的磷酸化和失活导致Rig-Ihep−/−小鼠中的胆固醇积累和肝脏脂肪变性被明显抑制。后续的分析发现,肝脏RIG-I与HMGCR上游的激酶AMPKα组成型结合,从而抑制HMGCR磷酸化。
考虑到翻译后修饰对蛋白质互作的重要性,研究者进一步检查了甲基化和乙酰化是否影响RIG-I与AMPKα的结合。有意思的是,只有模拟去甲基化的K18A和K146A突变体显著抑制RIG-I-AMPKα结合,而模拟单甲基化的K18M和K146M突变体则增强了它们的结合(此处使用的两种突变型小鼠模型均由赛业生物提供),表明RIG-I的K18和K146组成型甲基化负责其与AMPKα的相互作用。研究者认为,甲基化RIG-I与AMPKα的组成型结合抑制了HMGCR磷酸化,从而增强了HMGCR酶活和胆固醇合成。
3、RIG-I的表达与人类肝癌发生有关
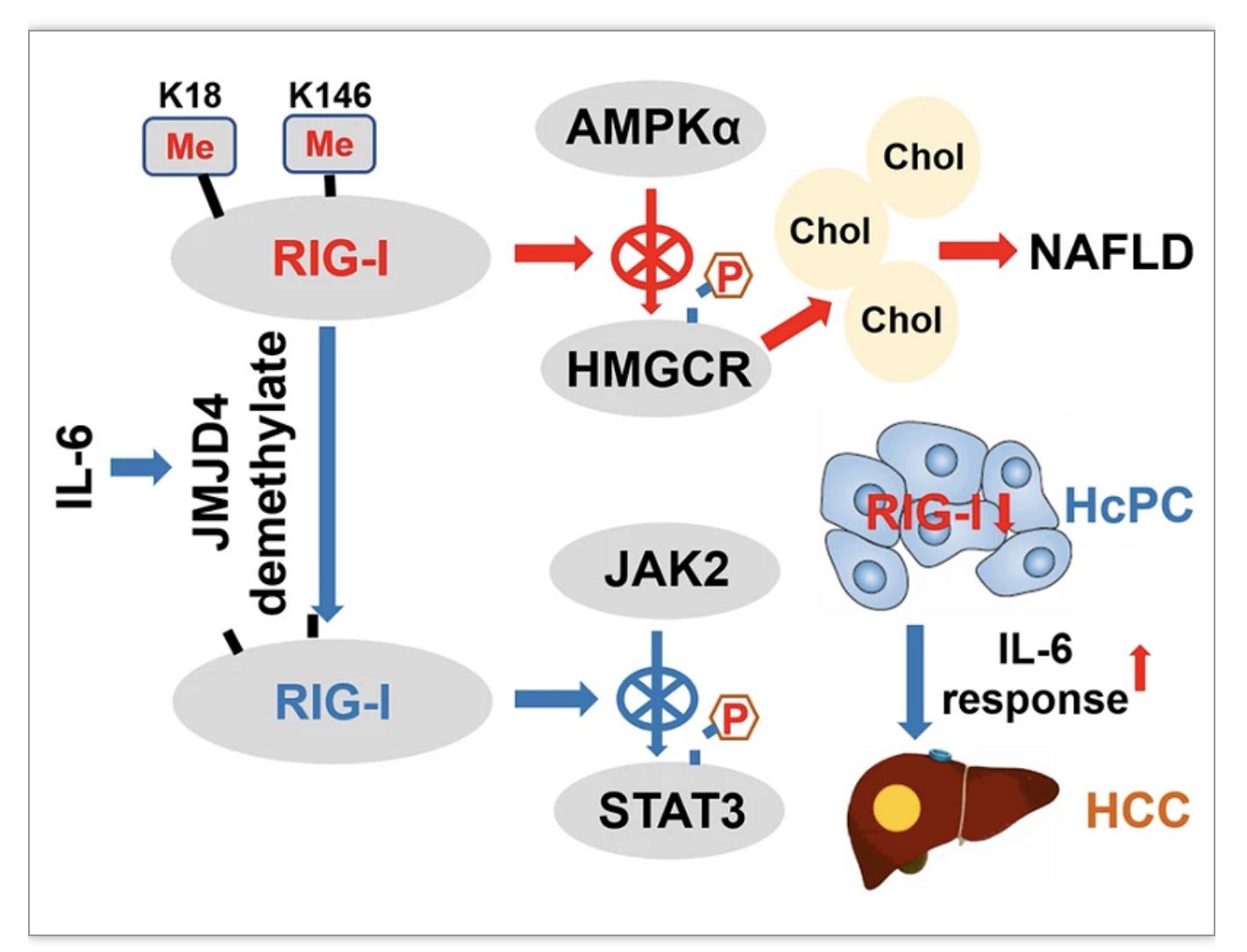
图3 RIG-I在肝癌发生中起作用的示意图[1]
