18101298214
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
首页
文献资料
肯尼迪病(KD)——机理研究
发布日期:2023/9/11 12:00:00
临床特点
肯尼迪病
(Kennedy’s disease,
KD
)又称为脊髓延髓肌萎缩症(Spinal and bulbar muscular atrophy, SBMA),是一种X连锁、成人发病型运动神经元疾病,其特征是延髓和四肢肌肉的缓慢进行性无力。肯尼迪病主要影响成年男性,估计本病的发病率(1~2)/10万,相当多的患者可能被误诊为其他神经肌肉疾病,包括肌萎缩侧索硬化症。
世界各地有不同种族背景的SBMA患者报道。
一般为30~60岁发病,疾病初期多为非特异性症状
,如姿势性震颤和肌肉痉挛。姿势性震颤多出现在患者肢体无力前数年甚至数十年前,值得引起临床医生重视,有研究发现其原因可能与临床下的感觉障碍和运动单位减少有关。早期临床症状包括面部收缩震颤或言语不清,多有肌束震颤,尤其是口周和舌头。
虽然目前认为肯尼迪病为下运动神经元病,但也有研究表明,肯尼迪病患者存在轻度认知功能障碍,表现为言语流畅性障碍、概念形成异常以及记忆力下降。部分肯尼迪病患者表现出雄激素不敏感的迹象,如乳房发育、睾丸萎缩、勃起障碍、生育能力降低。女性携带者通常无症状。体格检查发现以下运动神经元体征为主,可见轻度的肌肉萎缩、束颤,肌力轻度减退,肢体近端更明显,腱反射减弱或消失和感觉缺失。大多数患者血清中的肌酸激酶水平升高,部分合并高血压、高血脂症、轻度肝功能异常、葡萄糖耐受不良等。
肯尼迪病患者电生理检查以广泛的慢性神经源性损害为最主要改变,多伴感觉和运动神经传导异常,且感觉异常比运动异常更多见
[1]
。
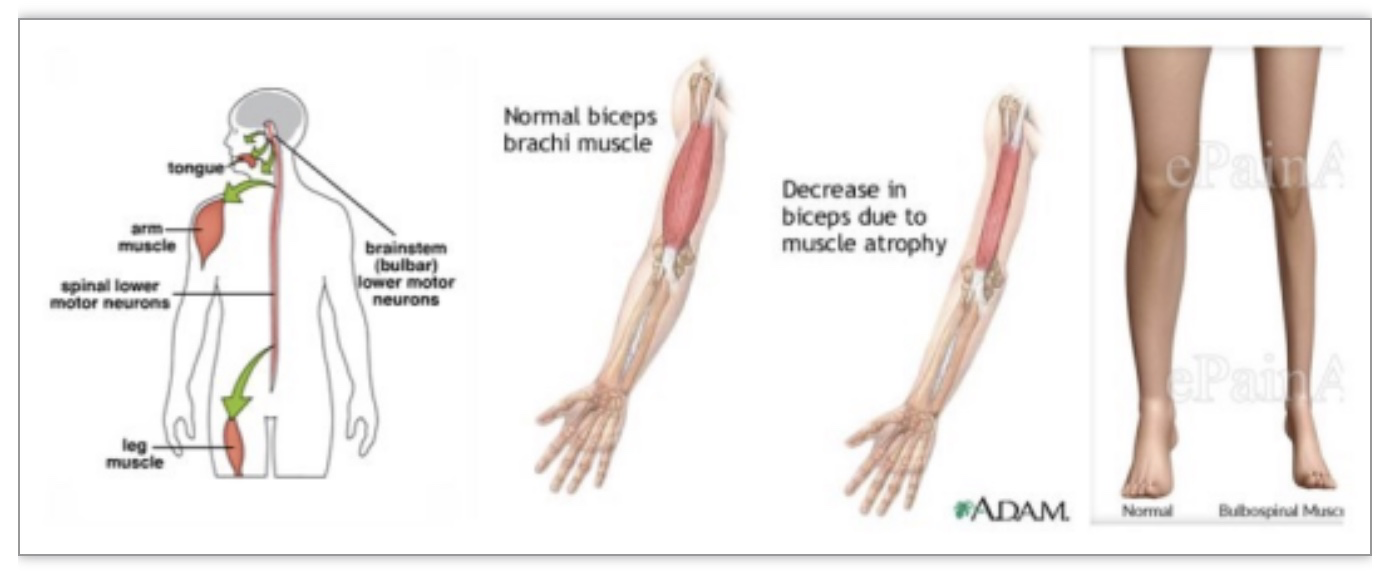
图1 肯尼迪病的临床特点[2]
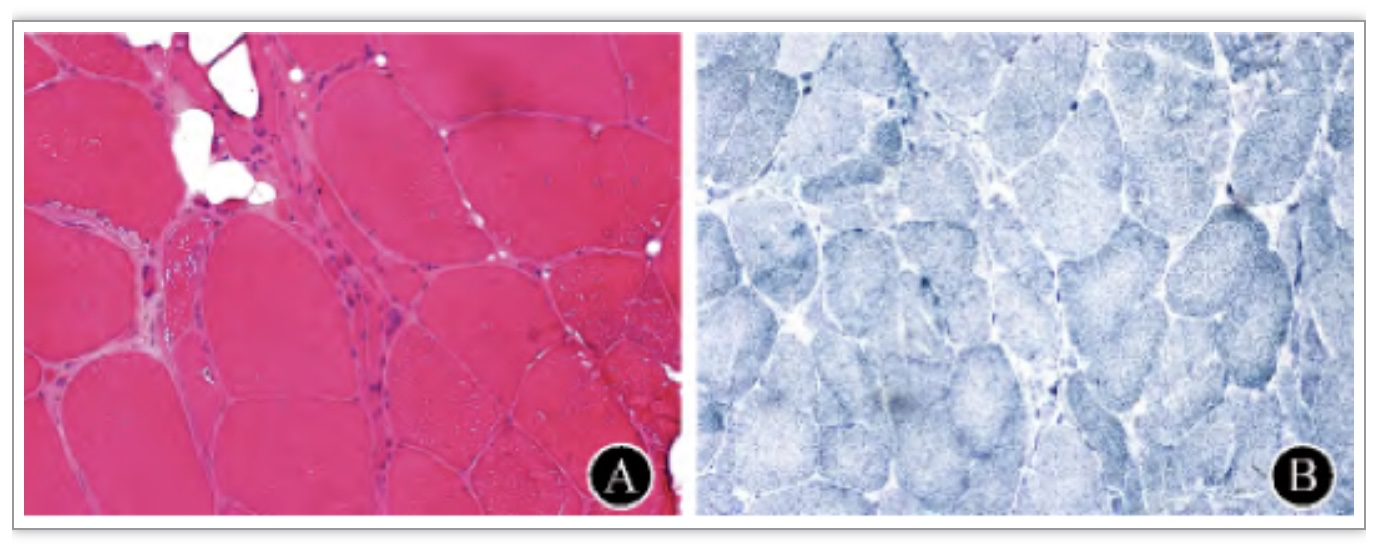
图2 肌肉病理检查 A:患者肌肉活检HE染色显示肌纤维呈重度萎缩,萎缩纤维呈束状分布,间质脂肪增多,未见炎细胞浸润;B:肌肉活检NADH染色显示Ⅰ型纤维成组化现象[3]
动物模型
有学者为了研究致病蛋白AR的特定配体——睾酮在SBMA中的作用,在巨细胞病毒增强子和鸡来源的β-actin启动子的控制下,
获得了表达含有97个CAGs的全长人AR的转基因小鼠,该模型不仅再现了神经功能障碍,而且还再现了与性别的表型差异,这是SBMA的一个特殊特征
[4]
。
又有学者鉴定了两个跨越AR基因整个长度的重叠的BACs,通过重组策略,将两个BAC融合,创建了一个AR BAC结构,除了所有8个AR外显子外,还包括DNA50到第一个AR外显子的50kb和DNA30的30kb到最后一个AR外显子,通过这种重组方法,该学者还引入了一个121CAG重复通道,并在AR外显子1两侧设计了两个loxP位点,构建了一个浮动的AR CAG121 BAC(BAC fxAR121)转基因结构体,然后获得了BAC fxAR121转基因小鼠,经过表达发现与YAC CAG100(YAC AR100)小鼠的hAR RNA、mAR内源性蛋白水平表达水平相当[5]。有学者通过此模型小鼠研究确定了肌肉是突变AR毒性的一个部位,并建议靶向该组织中的突变蛋白表达作为治疗该疾病的一种方法[6]。
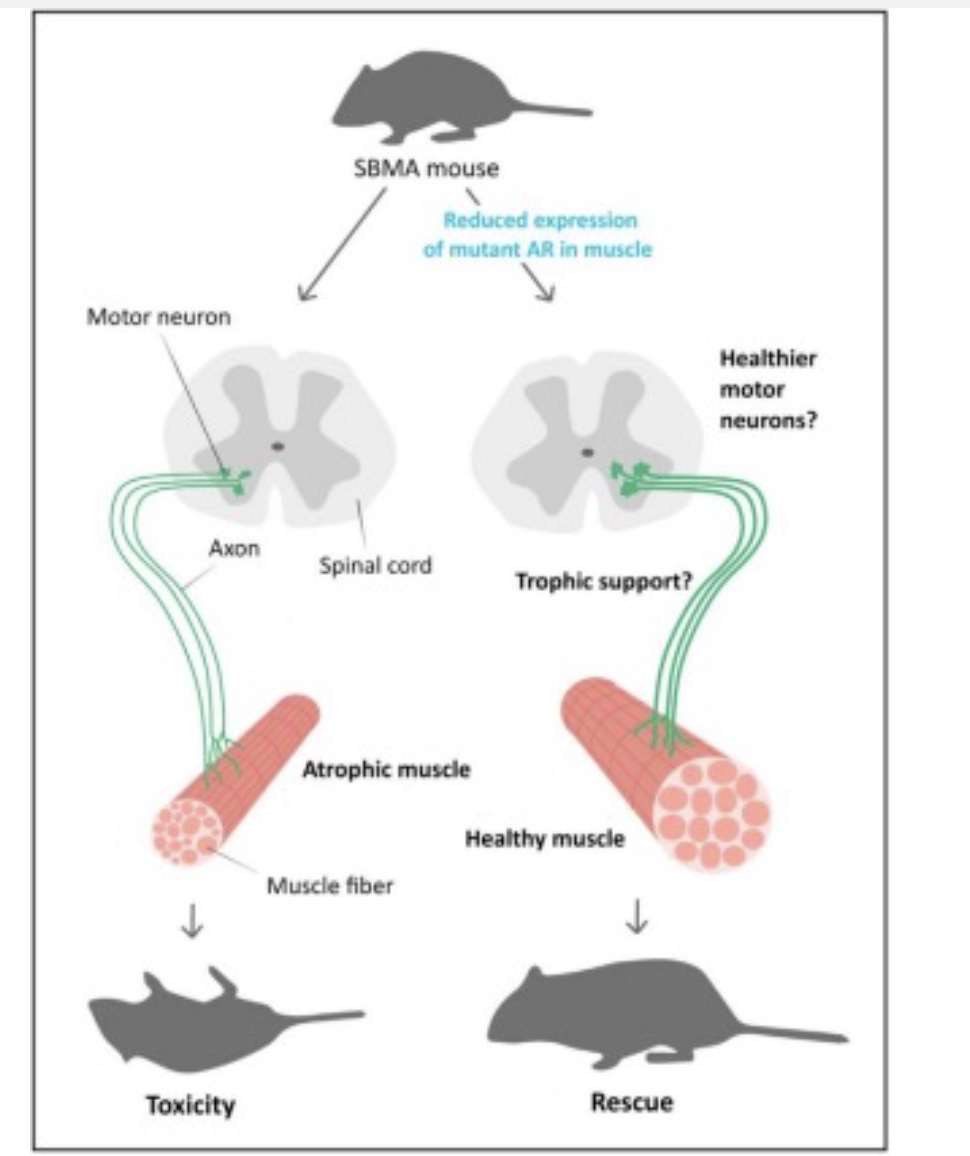
图3 减少肌肉中突变体AR的表达在SBMA小鼠模型中具有有益的作用[6]
治疗方式
●
对症治疗
对症治疗有助于缓解震颤、内分泌异常、肌肉痉挛、呼吸衰竭、吞咽困难等症状。
对已确诊为SBMA的患者应进行长期随诊观察。对于痛性痉挛,镁剂,替扎尼定、巴氯芬、加巴喷丁、丙戊酸钠,卡马西平等均可选用。若患者存在糖尿病,则按照现行诊疗原则进行治疗。若患者因吞咽困难出现营养不良,可行经皮内镜胃造瘘;对于小部分出现呼吸功能障碍的患者,无创正压机械通气可以改善患者症状;若患者晚期出现呼吸功能衰竭,必要时可根据患者意愿决定是否行机械辅助通气[7]。
●
特异性治疗
对于包括肯尼迪病在内的多聚谷氨酰胺疾病,多个机制可能参与神经元功能障碍和最终的细胞死亡。它们包括:错误折叠导致疾病的蛋白质功能改变;由突变型蛋白从事有毒性的蛋白质的相互作用;形成有毒的寡聚物;转录失调;线粒体功能障碍导致受损的生物能学和氧化应激;受损的轴突运输;异常的神经信号,包括兴奋性毒性;细胞蛋白质稳态受损;RNA毒性。虽然这些分子机制都有可能作为治疗靶点,
但因为肯尼迪病的致病机制尚未阐明,故目前仍没有标准的治疗方案,现有方案大都只停留在动物模型实验阶段。
在这些治疗方法中:
1.雄激素剥夺已经应用到临床试验
。一些动物实验已表明,
雄性肯尼迪病小鼠的组织病理学发现AR蛋白表达水平与血液中睾酮水平呈正相关
。
为了支持这一观点,在肯尼迪病的小鼠模型中,手术去势显示反向运动功能障碍。类似的结果也在亮丙瑞林的临床前研究中得到。亮丙瑞林是一种促黄体激素释放激素衍生物,可抑制垂体释放促性腺激素,抑制睾丸释放睾酮。这种药物已被用于各种性激素依赖性疾病,包括前列腺癌、子宫内膜异位症。在雄性肯尼迪病小鼠,亮丙瑞林成功抑制了致病的AR核积聚,导致神经肌肉的表型明显改善。亮丙瑞林的雄激素阻断作用也由前列腺和精囊的重量减少得到了进一步证实。
与对照组小鼠相比,亮丙瑞林治疗的肯尼迪病小鼠表现出更长的寿命、较大的尺寸、更好的运动性能
。
亮丙瑞林的2期临床试验显示,在亮丙瑞林治疗大约2~4周后,人血清睾酮水平下降到了手术去势水平。与安慰剂组比较,这种药物治疗的患者在阴囊皮肤活组织检查(活检)显示突变的AR积累减少,吞咽功能增强。接受亮丙瑞林治疗的患者尸检结果表明,雄激素剥夺抑制了脊髓和脑干运动神经元AR的核积。这些观察结果表明,亮丙瑞林通过给药抑制了突变体AR在肯尼迪病的神经肌肉毒性蓄积。
2.AR共同调节因子
也作为替代治疗目标,因为它们控制细胞AR的功能和分布。从生姜、咖喱中萃取的化合物5-羟基-1,7-双(3,4-二甲氧基苯基)-1,4,6-heptatrien-3-1(AsC-J9)可扰乱AR和其共同调节因子的相互作用。
近期研究发现AsC-J9可选择性降解AR蛋白并减少其在细胞内积聚,使AR-97O转基因小鼠的运动能力得到显著改善,平均生存期由28周增至39周,且AsC-J9治疗后的肯尼迪病小鼠,血清睾酮水平基本正常,性功能和生育能力显著改善。
3.细胞防御机制的激活
是肯尼迪病另一个有希望的治疗方法。在肯尼迪病的小鼠模型中,Hsp是一种应激诱导的分子伴侣,可分为不同的家族:Hsp100、Hsp90、HspT0、Hsp60、Hsp40和小Hsps。
在肯尼迪病的小鼠模型中,Hsp在多聚谷氨酰胺疾病中(包括肯尼迪病)的高水平表达可抑制异常蛋白的毒性积聚,并可通过多种途径阻止细胞死亡。
故通过药物诱导方法增加其表达水平,很有可能成为治疗肯尼迪病和其他多聚谷氨酰胺疾病的一种新方法。替普瑞酮(GA)在多种组织中都可强烈诱导Hsp的表达。予肯尼迪病转基因小鼠口服GA后可显著上调HspT0在中枢神经系统中的表达水平,并可抑制致病AR蛋白在细胞核内的积聚,从而显著改善神经肌肉相关的症状表现。另一方面,在肯尼迪病中,抑制Hsp90已被证实通过激活泛素-蛋白酶体系统抑制神经变性。
在肯尼迪病的细胞和小鼠模型中,用一种强效的Hsp90抑制剂-17-烯丙基氨基格尔德霉素(17-AG)进行治疗,促进了致病AR蛋白酶体的降解。
4.转录失调
是用于治疗干预的另一个靶点。抑制组蛋白去乙酰化酶(HDAC)活性可增加组蛋白乙酰化,随之增加基因的转录水平,HDAC抑制剂已被认为在多聚谷氨酰胺疾病中有治疗作用。丁酸盐是被发现的第一个HDAC抑制剂,
在肯尼迪病小鼠模型中,丁酸钠通过口服给药,可上调神经组织中组蛋白乙酰化的表达,改善模型的症状和病理表型
[8]
。
上一篇:
解析RIG-I在肝癌发生中的作用
下一篇:
发明高通量高稳定性的拉曼流式细胞术pDEP-DLD-RFC
已经到最底了
技术支持:
库价化学
Copyright © 2024北京螽斯羽生物有限公司 备案号:
京ICP备2023018288号-1