
肌肉减少症(sarcopenia,简称肌少症)是指与增龄相关的进行性、全身肌量减少和/或肌强度下降或肌肉生理功能减退为特征的综合征[1]。肌少症主要表现为老年人机体活动能力和肌肉功能下降,导致跌倒和骨折风险增加。肌少症已成为老年人群致残、致死的主要原因之一,给当今人口老龄化的社会造成严重负担。然而,目前对肌少症的认识尚在起步阶段,特别是对其发病机制的研究尚不深入,临床上尚缺乏有效的药物治疗。因此,需要对肌少症的发病机制进行深入研究,寻找更多新的治疗靶点。该研究首先在成功构建自然衰老肌少症小鼠模型基础上发现,肌少症小鼠呈现以循环及局部骨骼肌TNF-α水平升高为代表的慢性低度炎症反应状态;并且骨骼肌细胞发生了GSDME介导的细胞焦亡伴凋亡信号分子caspase-8、caspase-3的激活。该研究进一步在细胞水平上深入探索了TNF-α调控骨骼肌细胞焦亡及介导骨骼肌细胞发生细胞凋亡与焦亡间相互转换的分子机制,为深入认识肌少症的发病机制及其治疗提供了新思路。

作者首先设立了三组不同年龄段小鼠:20周龄小鼠代表年轻组、60周龄小鼠代表中年组、88周龄小鼠代表老年组。通过观察对比了三组小鼠的肌力、肌肉质量、腓肠肌HE染色及电镜表现,成功构建了自然衰老的小鼠老年肌少症模型。在此基础上,作者发现老年小鼠血清TNF-α水平及腓肠肌TNF-α表达水平均显著高于年轻组和中年组。此外,老年组小鼠腓肠肌高表达GSDME的N端水解段(GSDME-N),而不表达GSDMD的N端(图1A,1B);免疫荧光提示老年组小鼠腓肠肌细胞高表达GSDME(图1D);TUNEL染色同样提示老年组小鼠腓肠肌显著性阳性反应(图1D)。作者同时发现老年组小鼠腓肠肌caspase-8、caspase-3被活化(图1A,1C,1D)。以上结果提示老年肌少症小鼠腓肠肌发生了GSDME介导的细胞焦亡伴随凋亡信号分子caspase-8及caspase-3的活化。
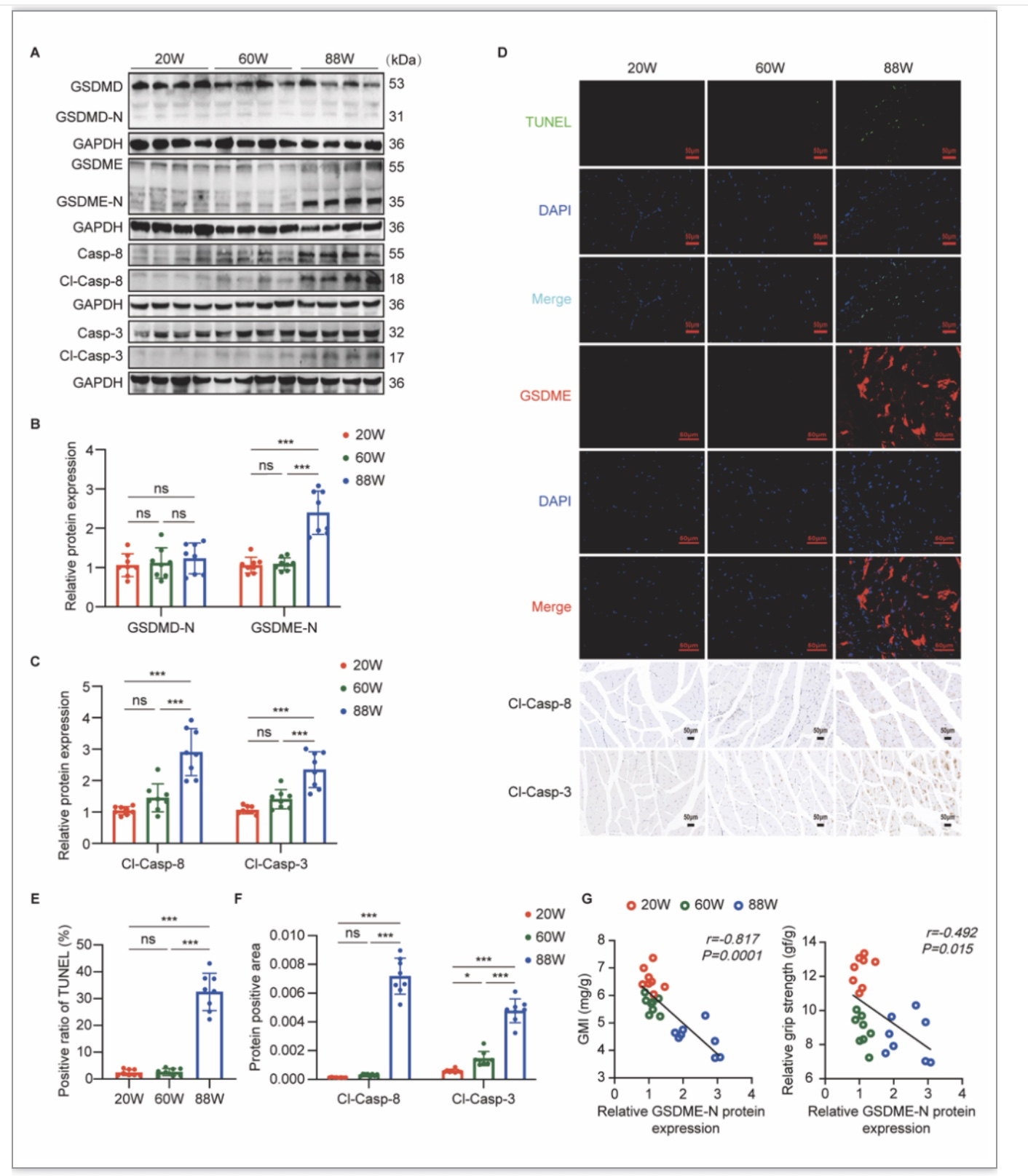
图1 老年肌少症小鼠腓肠肌发生GSDME介导的细胞焦亡伴caspase-8/3的活化
(图源:Wu J et al., Cell Death Discov, 2023)
作者接着以TNF-α处理小鼠C2C12肌管细胞,通过摸索干预浓度及时间,确定了TNF-α诱导C2C12肌管细胞发生GSDME介导的细胞焦亡的有效浓度与时间。在此基础上,作者发现TNF-α亦可诱导C2C12肌管细胞caspase-8及caspase-3的活化(图2A,图3A)。由于在TNF-ɑ诱导细胞死亡的信号通路中,caspase-8是介导细胞发生凋亡或坏死样凋亡的关键分子。当caspase-8被活化时可水解坏死样凋亡通路的关键分子RIPK1、RIPK3从而阻断坏死样凋亡,使细胞发生凋亡[2]。因此,当TNF-ɑ与caspase-8抑制剂Z-IETD-FMK联合干预细胞时,细胞的凋亡通路虽被阻断了,但RIPK1、RIPK3未被抑制进而使细胞转向坏死样凋亡通路[2]。为了阻断坏死样凋亡通路对研究中观察细胞死亡的干扰,作者接下来对TNF-α刺激下的C2C12肌管细胞同时加用了Z-IETD-FMK和RIPK3抑制剂GSK’872进行干预。作者发现Z-IETD-FMK和GSK’872可逆转TNF-ɑ所诱导的细胞焦亡的发生(图2B、2E、2C);并且也抑制了TNF-ɑ对caspase-3的活化作用(图2B)。此外,作者对TNF-α刺激下的C2C12肌管细胞同时加用caspase-3抑制剂Z-DEVD-FMK干预后,亦发现Z-DEVD-FMK可逆转TNF-ɑ所诱导的细胞焦亡的发生(图3B-3E)。这些结果表明TNF-α通过caspase-8/caspase-3诱导肌管细胞发生GSDME介导的细胞焦亡。
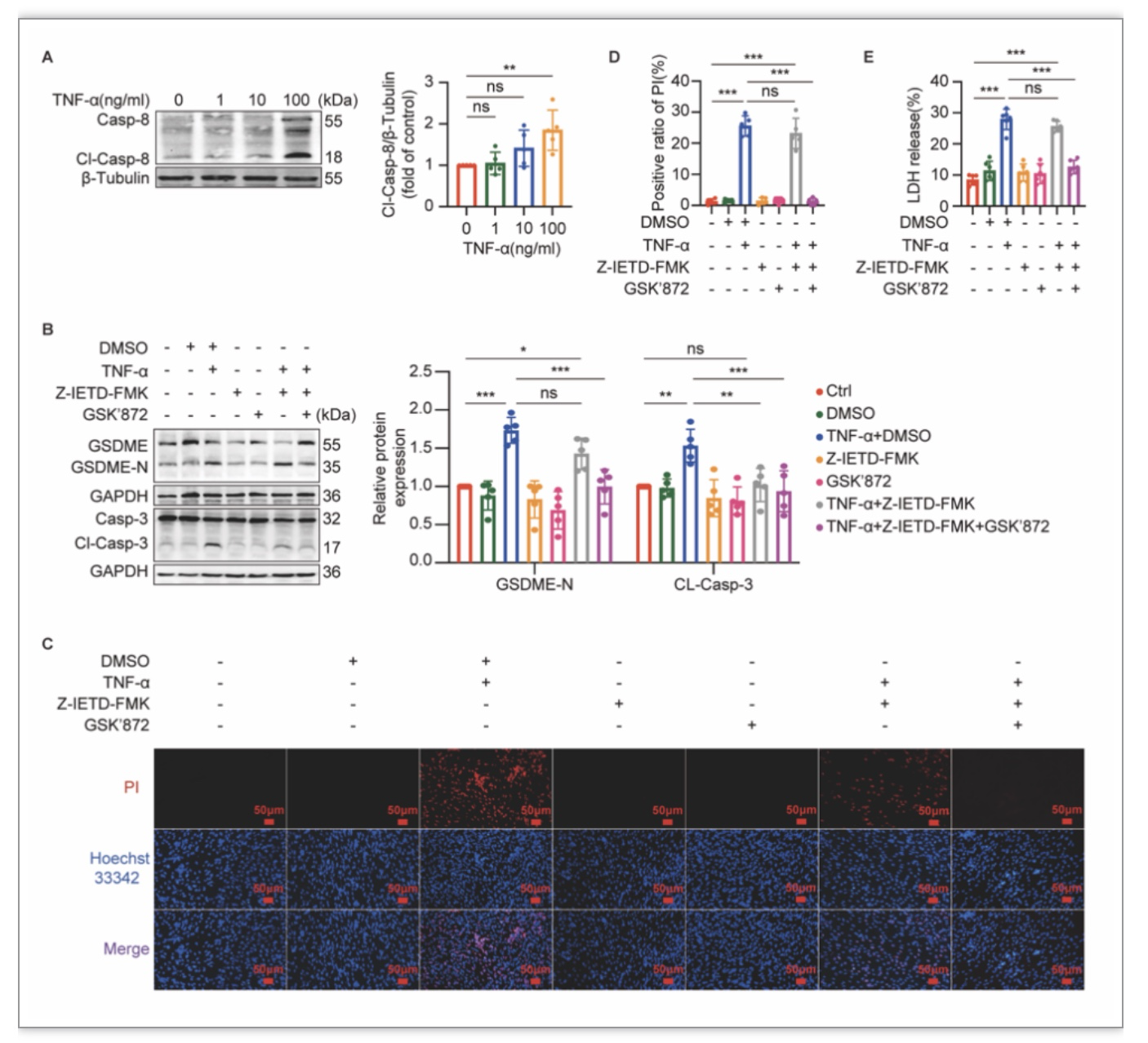
图2 TNF-α通过激活caspase-8诱导C2C12肌管细胞焦亡的发生
(图源:Wu J et al., Cell Death Discov, 2023)
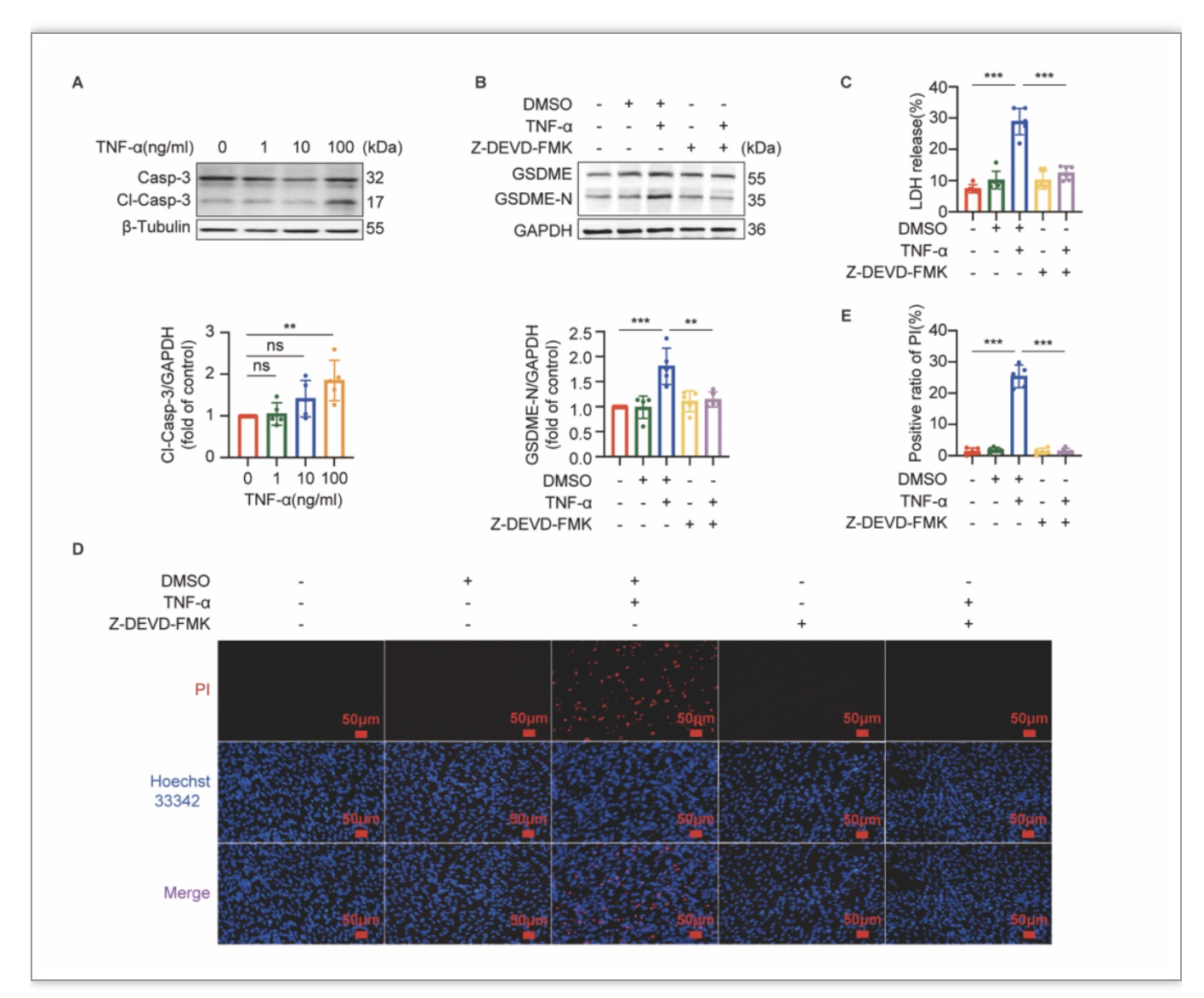
图3 TNF-α通过激活caspase-3诱导C2C12肌管细胞焦亡的发生
(图源:Wu J et al., Cell Death Discov, 2023)
通常情况下,TNF-ɑ可通过细胞内TNF复合体IIa或TNF复合体IIb的组装而激活caspase-8[3]。然而尚不清楚TNF-ɑ在C2C12肌管细胞内究竟是通过何种复合体激活caspase-8以最终诱导细胞焦亡的发生。既往研究都将TNF-ɑ与蛋白质合成抑制剂如放线菌酮(CHX)同时刺激细胞以诱导胞内TNF复合体IIa形成;而TNF-ɑ与转化生长因子-β活化激酶1抑制剂(TAK1i)即5Z-7-Oxozeaenol同时刺激细胞则可诱导胞内TNF复合体IIb形成[3]。因此,作者进一步以TNF-ɑ+ CHX和TNF-ɑ+ TAK1i分别刺激C2C12肌管细胞以诱导胞内TNF复合体IIa和IIb的形成。TNF-ɑ+ TAK1i在刺激肌管细胞6h即可诱导细胞焦亡的发生(图4A),而TNF-ɑ+ CHX则在刺激肌管细胞24h出现细胞焦亡的发生(图4B)。在刺激肌管细胞6h时,与TNF-α+CHX刺激的肌管细胞相比,TNF-ɑ+ TAK1i刺激的肌管细胞表达GSDME-N的水平显著增加(图4D);而在刺激肌管细胞24h时,两者所诱导的GSDME-N的水平未见显著性差异(图4C)。同样地,在刺激肌管细胞6h时,与TNF-α+CHX刺激的肌管细胞相比,TNF-ɑ+ TAK1i可显著性诱导肌管细胞caspase-8的活化(图4D)。这些结果表明TNF-ɑ在刺激肌管细胞时可能更倾向于诱导胞内TNF复合体IIb的组装,进而激活caspase-8并诱导GSDME介导的细胞焦亡的发生。
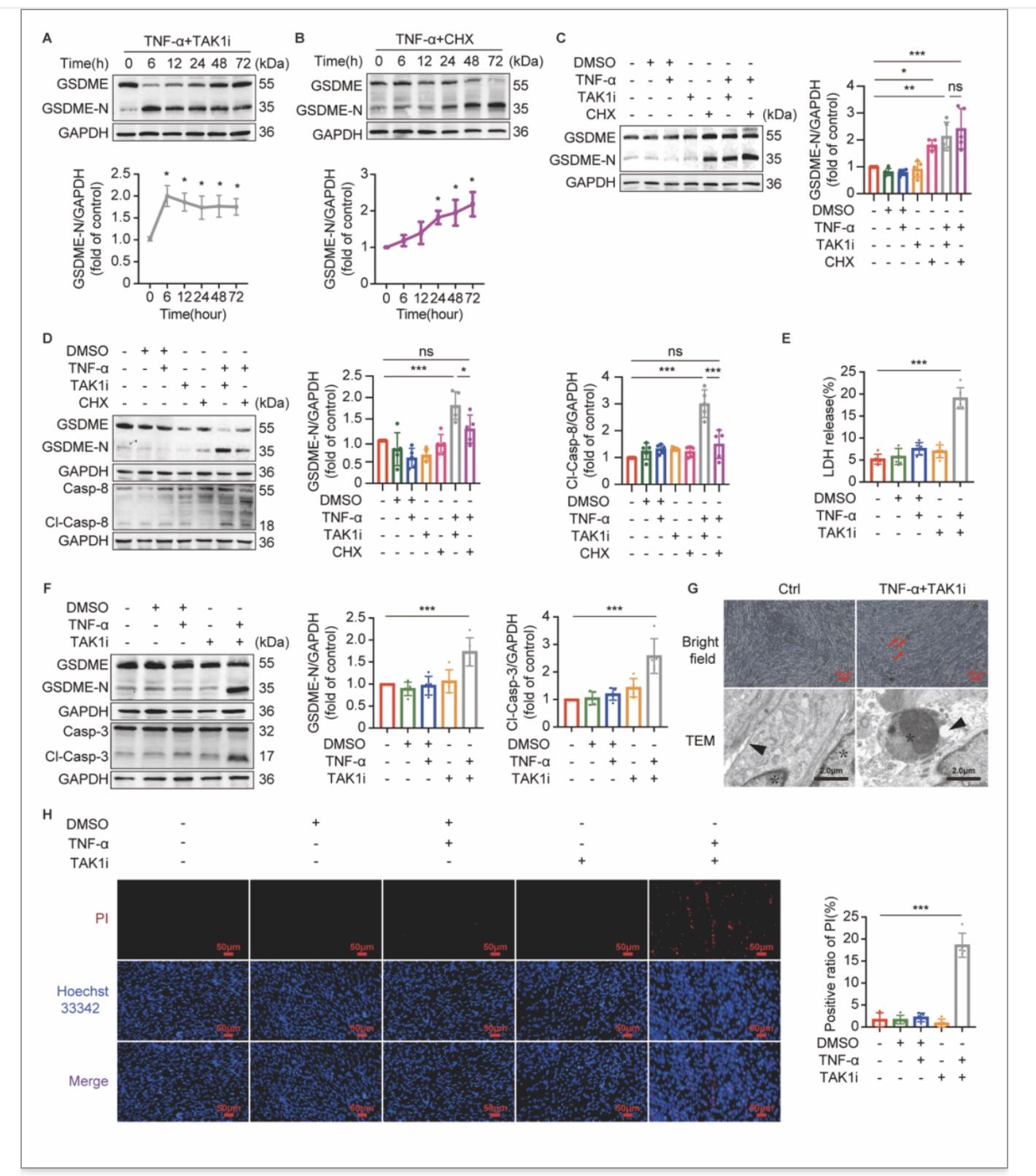
图4 TNF-α通过在肌管细胞内形成TNF复合体IIb而诱导caspase-8/caspase-3/GSDME介导的细胞焦亡的发生
(图源:Wu J et al., Cell Death Discov, 2023)
基于既往肌少症的体外研究,作者以肌球蛋白重链1(MHC1)作为体外研究肌少症的标志物[4]。作者首先以TNF-ɑ和TNF-ɑ+ TAK1i分别刺激肌管细胞,均发现MHC1表达水平显著下降(图5A-5C),证实TNF-ɑ可在体外诱导肌少症的发生。接着,作者再次对TNF-α+ TAK1i刺激下的肌管细胞同时加用了Z-IETD-FMK和GSK’872进行干预。作者发现Z-IETD-FMK和GSK’872可逆转TNF-ɑ所诱导的肌少症的发生(图5D)。同样地,作者对TNF-α+ TAK1i刺激下的肌管细胞同时加用Z-DEVD-FMK干预后,亦发现Z-DEVD-FMK可逆转TNF-ɑ所诱导的肌少症的发生(图5E)。接着,作者以沉默GSDME的慢病毒载体(LV-shGSDME)感染C2C12成肌细胞并成功诱导分化为肌管细胞(图5G)。最后,作者以TNF-ɑ+ TAK1i刺激LV-shGSDME肌管细胞,发现沉默GSDME表达可逆转TNF-ɑ+ TAK1i所诱导的细胞焦亡(图5H,5J),并且MHC1的表达水平也显著增加(图5I)。这些结果表明TNF-ɑ/TNF复合体IIb/ caspase-8/caspase-3介导的细胞焦亡参与了肌少症的发生发展。

图5 TNF-ɑ/ TNF复合体IIb /caspase-8/caspae-3/GSDME信号通路介导的细胞焦亡促进肌少症的发生
(图源:Wu J et al., Cell Death Discov, 2023)
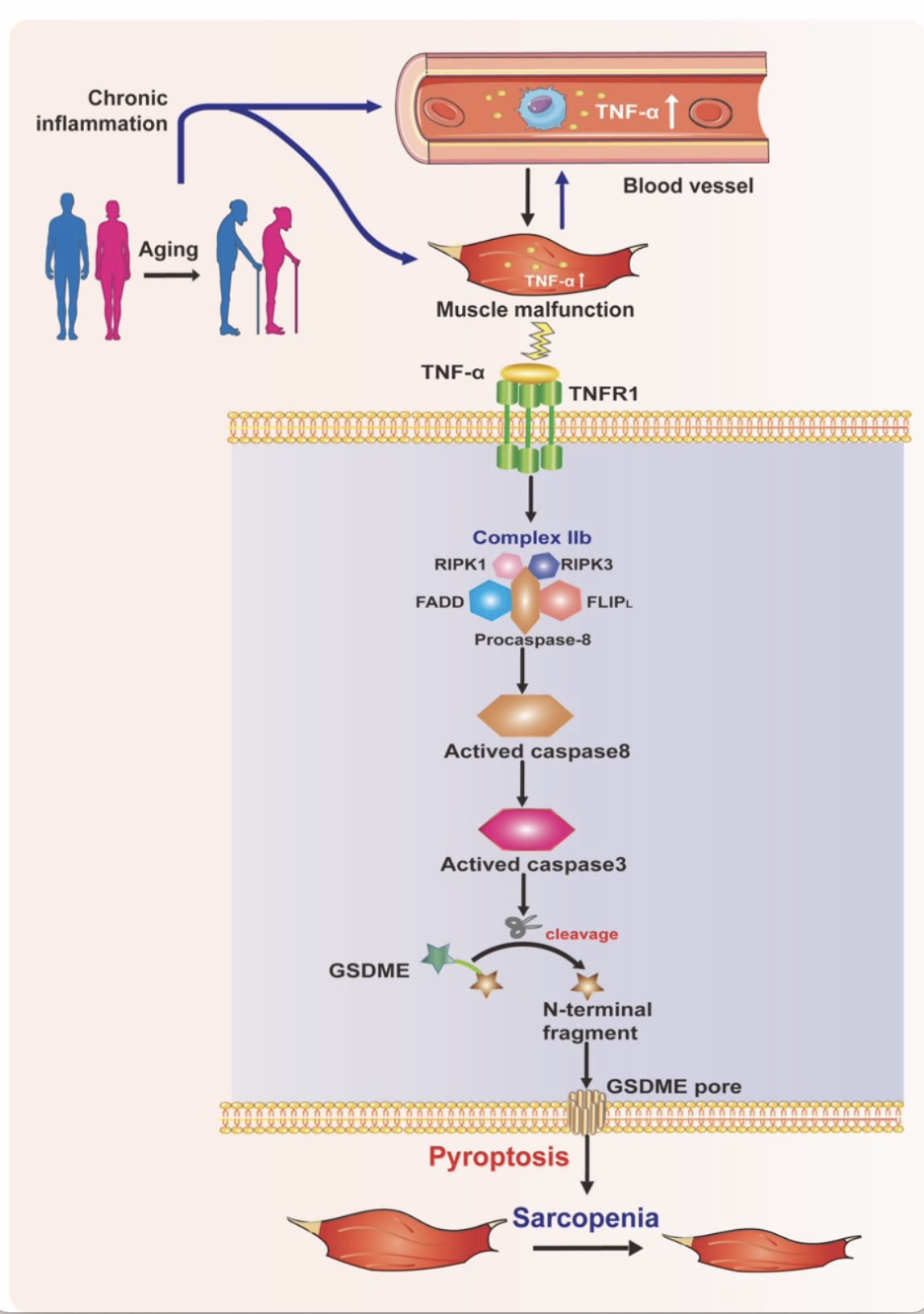
图6 TNF-α/ TNF复合体IIb/caspase-8/caspase-3/GSDME介导的细胞焦亡促进肌少症发生的机制图
(图源:Wu J et al., Cell Death Discov, 2023)
