抑癌基因TP53是人类肿瘤中最常发生突变的基因,一直是肿瘤学研究的重点和热点。肿瘤的发生发展与TP53突变引起的功能障碍密切相关。p53蛋白主要作为转录因子,调节多种细胞途径,如细胞周期阻滞、DNA修复、细胞凋亡和自噬等,并决定细胞在压力条件下是否死亡。多年来,一系列研究揭示了p53通路的复杂性,并延伸出其在代谢稳态、免疫微环境、肿瘤细胞干性等各种方面的功能。然而,突变的p53可以改变其DNA结合特性,破坏蛋白质的空间构象和热稳定性,并导致p53功能的紊乱。
TP53在肿瘤中的高频率突变和野生型TP53固有的抑癌功能使p53成为极具潜力的肿瘤治疗靶标。然而,由于p53蛋白质结构表面光滑,缺乏理想的药物结合口袋,导致恢复p53的抑癌功能十分困难。长久以来,p53被认为是一个“不可成药”的靶标,至今仍没有特异性靶向p53的药物获得批准上市。因此,十分有必要及时总结p53的最新研究进展,全面剖析野生型p53和突变型p53介导的下游信号通路在肿瘤中的功能,阐明p53蛋白的关键结构特征及其靶向治疗的研究现状,为实验室设计到临床开发的抗癌治疗提供重要参考。
作者首先对p53信号通路的各种分子机制以及p53突变如何影响肿瘤进展进行了系统的分析,进而讨论了p53蛋白的关键结构特征及其因致癌突变而失活的机制,随后深度解析了p53药物开发的进展和趋势,最后评述了p53抗癌治疗在临床开发中所遇到的挑战,并为未来发展方向进行了展望。

p53首次报导于1979年,p53蛋白被发现可以与致癌病毒SV40形成复合物[1],将克隆得到的p53转入到细胞可引起细胞癌变,所以最初普遍认为TP53是一种致癌基因[2,3]。但随着研究的深入,人们发现这一认识是错误的。首次发现的p53是突变的,在肿瘤细胞中,TP53基因也经常发生突变或丢失,而在正常细胞中,TP53基因是完整的。野生型p53不但不会致癌,而且在细胞培养环境下会抑制细胞癌变和肿瘤细胞生长。目前,TP53已成为研究最多的肿瘤抑制基因之一。
肿瘤抑制因子p53是抑制肿瘤发生和发展的主要关卡,p53作为一个序列特异性转录因子,能够结合基因组内特定的DNA序列并调节下游基因的转录,在调控细胞周期、DNA修复、凋亡和衰老等方面发挥重要作用[4]。正常情况下,细胞内p53蛋白水平非常低,这是由于负调控因子MDM2和MDMX严格控制p53的泛素化降解[5]。当细胞暴露于内外部压力如DNA损伤、缺氧、营养剥夺和癌细胞风险时,p53泛素化被抑制,细胞内p53蛋白水平迅速升高。稳定的p53在细胞核中形成四聚体,与目标DNA结合并调节基因转录[6]。p53通过转录激活凋亡和细胞周期相关的各种基因,阻止DNA突变或受损的细胞分化,阻止癌变细胞的产生。此外,一系列研究表明,p53还控制着许多“非经典”通路,包括代谢稳态、铁死亡、肿瘤细胞干性、自噬、衰老、肿瘤微环境等(图1)。
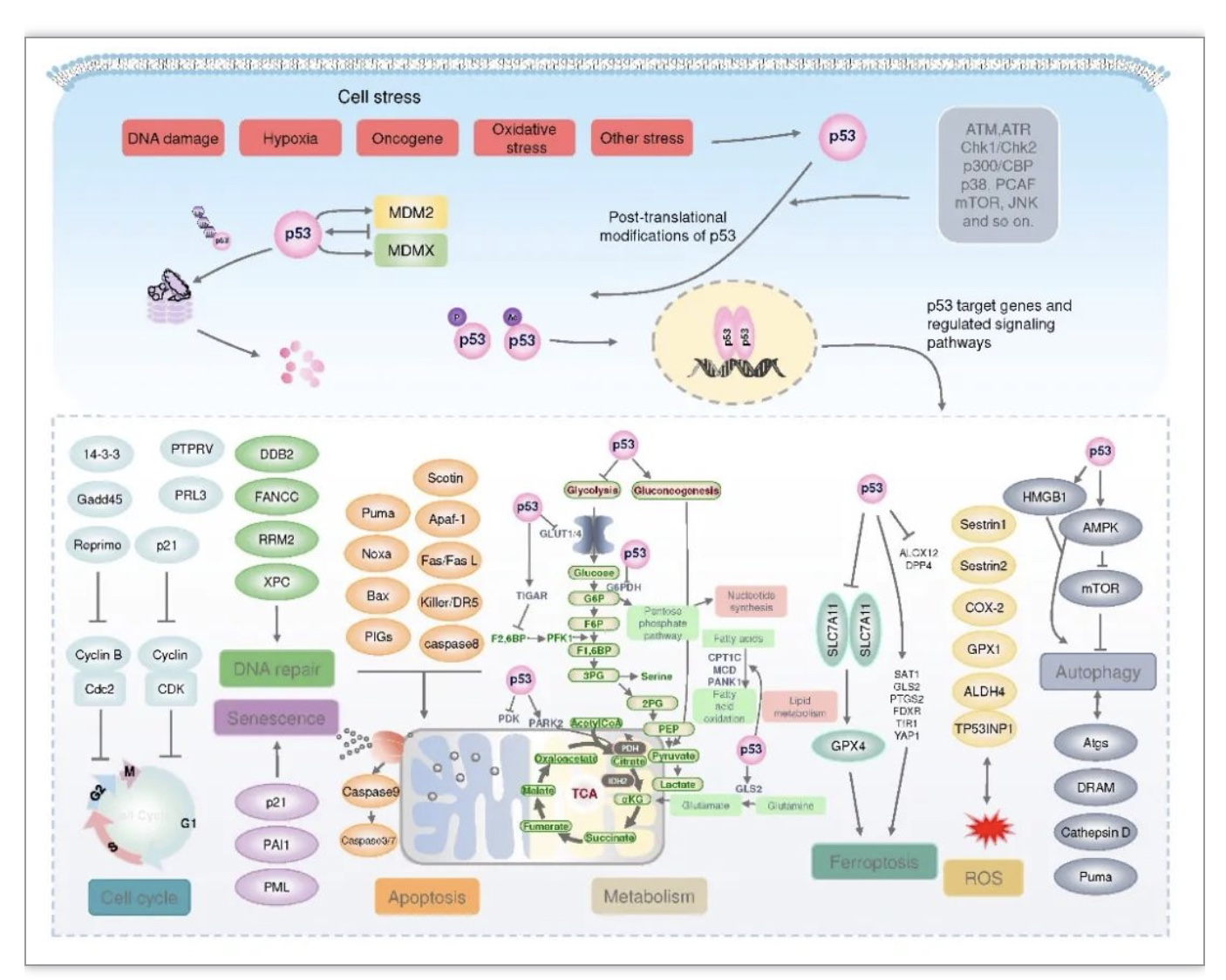
图1 p53信号通路
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
TP53基因在大多数肿瘤细胞中发生突变。不同人类肿瘤细胞的基因组测序显示,42%的病例携带TP53突变[7]。突变型p53失去肿瘤抑制功能,或结合野生型p53蛋白并抑制野生型p53功能,还可能转化为致癌蛋白。TP53突变为肿瘤发生提供了一个有利的环境。肿瘤细胞中TP53高频率突变可能是压力性选择的结果,这种压力有利于肿瘤细胞逃避监测,从而免于死亡。许多突变型p53蛋白比野生型p53蛋白更稳定,可以在细胞中积累。一些突变型p53蛋白可能具有与野生型蛋白完全不同的功能,这可能是由于靶基因谱的改变、突变的p53结构紊乱或不适当的蛋白质-蛋白质相互作用造成的(图2)。

图2 突变p53在肿瘤中的作用
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
p53突变的氨基酸位点大部分位于其DNA结合域(图3),通常分为结构突变体和DNA接触面突变体。结构突变体(如R175H, R249S, G245S, Y220C)降低了蛋白质的热稳定性,导致蛋白质在生理温度下不能正常折叠,失去与DNA结合的能力。DNA接触面突变体(如R273H/C, R248W)位于DNA核心结合区,导致p53与DNA结合异常。R175、G245、R249、R282、R248和R273是最常见的突变位点,因此被称为p53的“热点”突变。

图3 癌症中的TP53突变
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
p53是一种多结构域蛋白,由393个氨基酸组成。它包含一个N端活化结构域(TAD,残基1-61)、一个富脯氨酸结构域(PRD,残基64-92)、一个DNA结合结构域(DBD,残基96-292)及相连的四聚化结构域(TET,残基324-356)和一个C端调控结构域(CTD,残基364-393) (图3)。其中,DBD负责结合和识别特异序列的DNA,这是p53作为转录因子的生物学功能核心。此外,DBD还能与不同的蛋白质相互作用[8],协调而微妙地调节各种生物学功能(图4)。
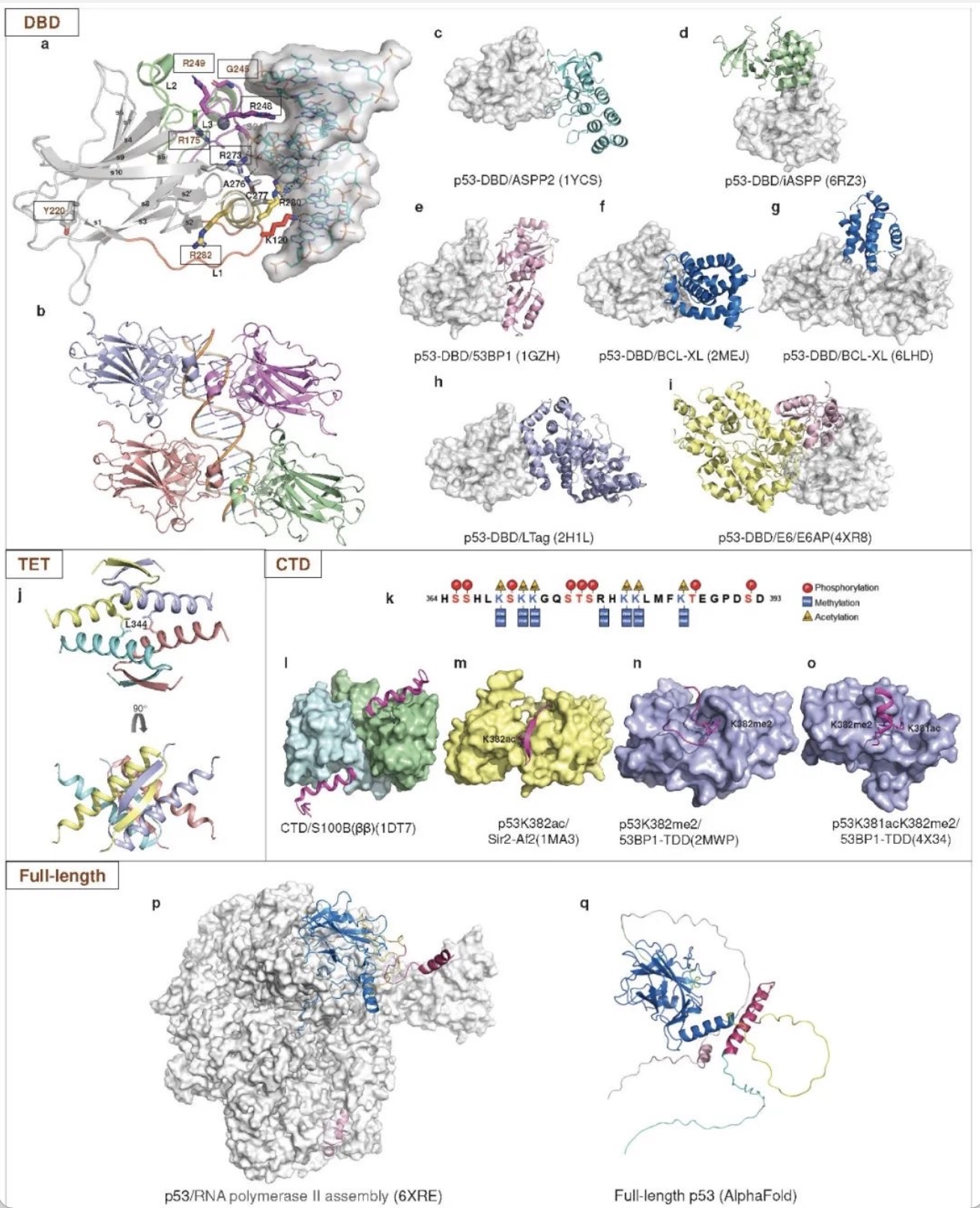
图4 p53的结构
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
肿瘤细胞中TP53的状态不同,需要的治疗策略也不同。对于具有野生型p53的肿瘤细胞,主要的方法是利用小分子化合物靶向p53的负调控因子MDM2/MDMX。MDM2抑制剂可有效解离p53/MDM2复合物,抑制MDM2介导的p53泛素化和降解,诱导肿瘤细胞中野生型p53的激活和积累。迄今为止,已有11种MDM2抑制剂进入临床试验阶段(图5)。然而,长期服用MDM2抑制剂可能导致TP53突变和MDMX表达增加,从而产生获得性耐药。此外,正常组织中p53的积累可能产生更大的毒性作用。因此,迫切需要开发MDM2/MDMX双靶点或特异性MDMX抑制剂,并对癌细胞精准使用MDM2抑制剂。
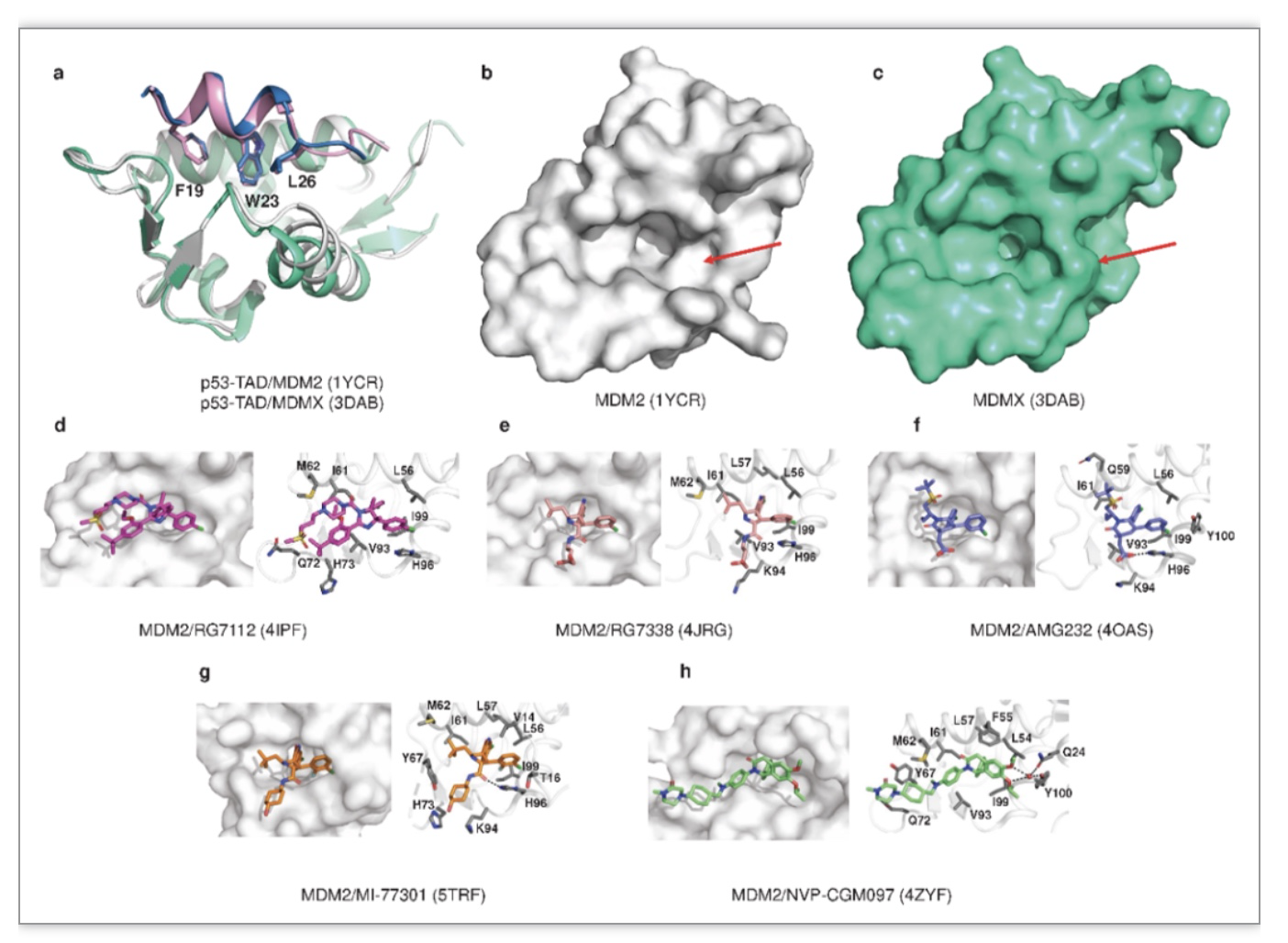
图5 MDM2与其小分子化合物的共晶结构
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
对于存在TP53错义突变的肿瘤,药物的研发主要集中于恢复突变体p53蛋白的野生型构象,从而恢复其肿瘤抑制功能。小分子化合物PRIMA-1和APR-246通过结合暴露在p53表面的半胱氨酸恢复突变体p53二聚体形成或DNA结合功能[9]。ATO[10]和PAT[11]靶向p53突变后产生的结构空腔中,帮助稳定p53空间结构,可以挽救大部分结构突变体p53的功能。ZMC1作为离子载体,将锌离子转移到细胞质,使细胞质锌离子浓度保持在适当范围内,从而重新激活突变体p53[12]。
p53中的Y220C突变是一个相对特定的突变。Y220远离p53与DNA结合的位点,导致p53表面形成疏水腔[13]。因此,Y220C的疏水口袋成为一个理想的药物结合靶点,在不干扰p53与DNA结合的情况下,帮助p53恢复正常的空间构象(图6)。
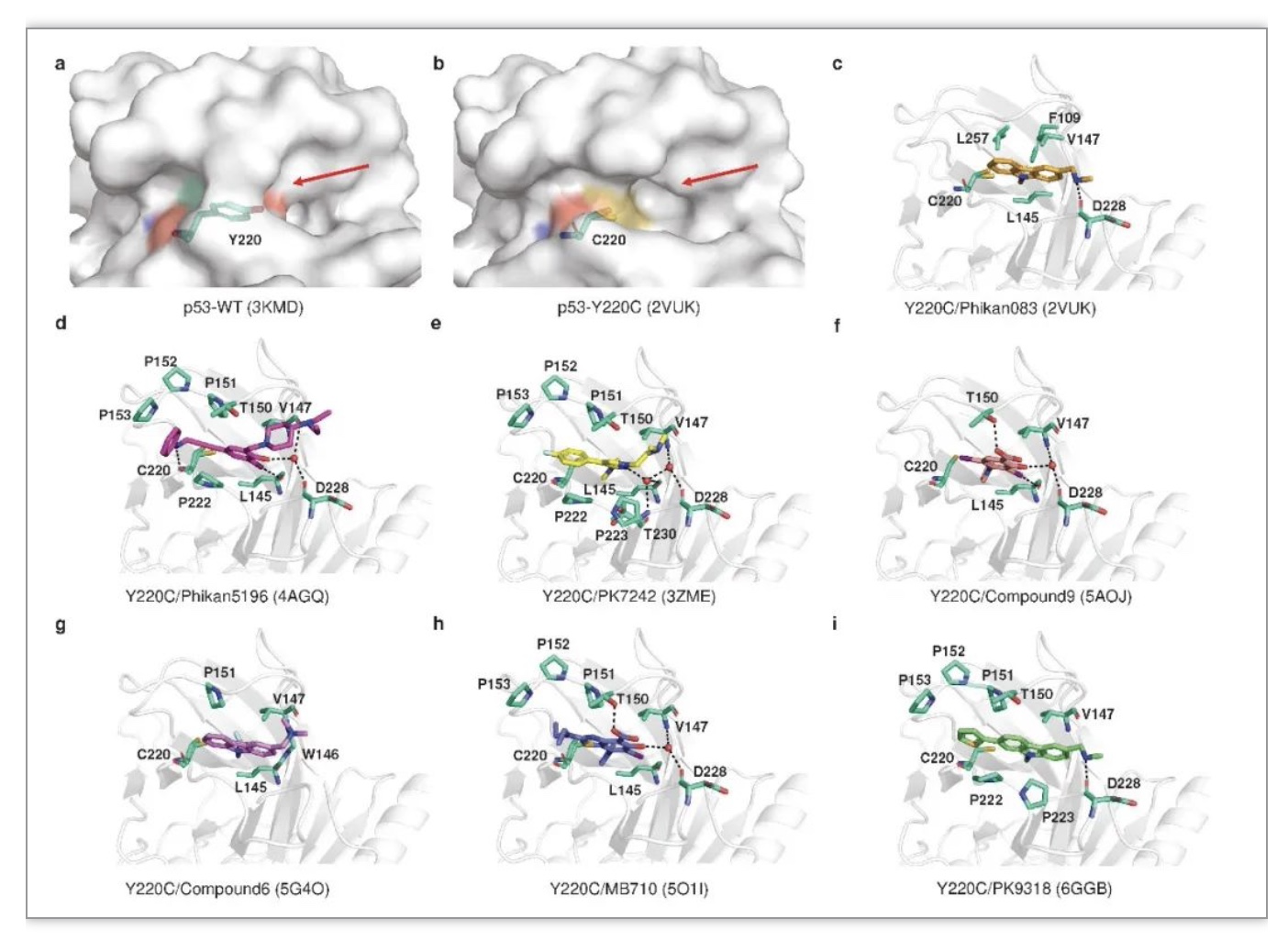
图6 p53突变体Y220C与小分子的共晶结构
(图源:Wang, H., et al., Signal transduction and targeted therapy, 2023)
p53是一种温度敏感蛋白,突变后其结构稳定性受到损害,暴露出包裹在p53疏水核心中的粘附序列,从而驱动p53聚集体的形成。p53聚集体剥夺了p53正常的DNA识别功能和促凋亡能力。已在多种肿瘤中发现由TP53突变引起的淀粉样蛋白聚集。ReACp53[14]、ADH-6[15]、LI/LH[16]和HDAC6/Hsp90抑制剂已被证明能有效抑制p53聚集并恢复p53的转录功能。
此外,靶向p53的多肽类药物、抗体类药物和mRNA,以及靶向p53的截短体和异构体等,都为肿瘤治疗提供了更多的选择。
开发靶向p53的药物过程中存在着重大挑战,缺乏既定的蛋白质再激活机制和缺乏药物结合口袋(Y220C除外)是两个主要原因。简单地利用化合物药物占据蛋白质的活性位点来抑制蛋白质的功能是相对容易的,然而如何利用化合物导致蛋白质功能的再激活是难以捉摸的。还有一些值得关注的问题是,TP53突变的耐药性、药物的脱靶效应,以及p53在正常组织中蓄积可能产生的毒副作用等,都使p53难以药物化。此外,TP53突变是异质性的,并不是所有的突变都相同。最后,单独的p53靶向治疗可能不足以治疗癌症。
该文章指出,后续p53靶向治疗的研究中有一些值得注意和考虑的因素。首先,鉴于TP53突变的异质性,一种药物万能的方法可能不适用于靶向TP53突变,不同的p53突变体可能需要不同的p53靶向药物。其次,联合治疗(如同时阻断MDM2-p53通路和p53-BCL-2通路)可能具有合成致死机制。最后,我们需要探索新的治疗方向,如靶向p53 mRNA、靶向紊乱结构域、靶向突变蛋白降解,以及使用CRISPR-Cas9进行基因组编辑等。随着CRISPR-Cas9技术的进步,使用CRISPR-Cas9纠正TP53突变可能成为未来一种有效的癌症治疗选择。
几十年来,针对p53的药物开发一直缺乏有效进展,p53曾被认为是一种不可药物的靶点。随着技术的进步,许多“不可成药”靶标也在逐渐变为药物靶标,如KRAS。我们有理由相信,针对p53的药物也会取得进展。鉴于TP53突变在人类癌症中的普遍存在,靶向p53的药物可能会带来癌症治疗的突破。

