
急性肾损伤(Acute Kidney injury, AKI)是肾小球滤过率(Glomerular Filtration Rate, GFR)迅速下降或排尿量减少为特征的临床综合征,具有较高的发病率和死亡率。在重症病房,约有50%的患者发生AKI,甚至发展成慢性肾脏病,明显的增加了住院患者的住院时间,增加医疗成本,成为一项重大的医疗卫生挑战[1]。AKI的病理生理机制复杂,且缺乏有效的靶向药物,因此进一步探索AKI新型的治疗靶点,对于预防、延缓AKI的发生发展具有重大的意义。
紧密连接(Tight Junctions, TJs)是锚定相邻肾小管上皮细胞 (Tubular Epithelial Cells, TECs)的主要结构之一,维持TECs的完整性。TJs的破坏可导致GFR下降和尿量的减少[2]。许多研究表明,AKI的发生与TJ蛋白的异常表达相关。然而通过各种不同的干预方式恢复TJ蛋白的表达,可改善AKI的肾功能。因此,对TJs 的深入探索,阐明TJs在AKI的发病机制,对于AKI新型的靶向药物的研发具有重要的指导价值。基于TJs在肾脏中的生物学功能和分布的特点,作者系统地阐述了TJs在AKI发病机制中的作用,并探讨了TJs可能作为AKI潜在的治疗靶点。

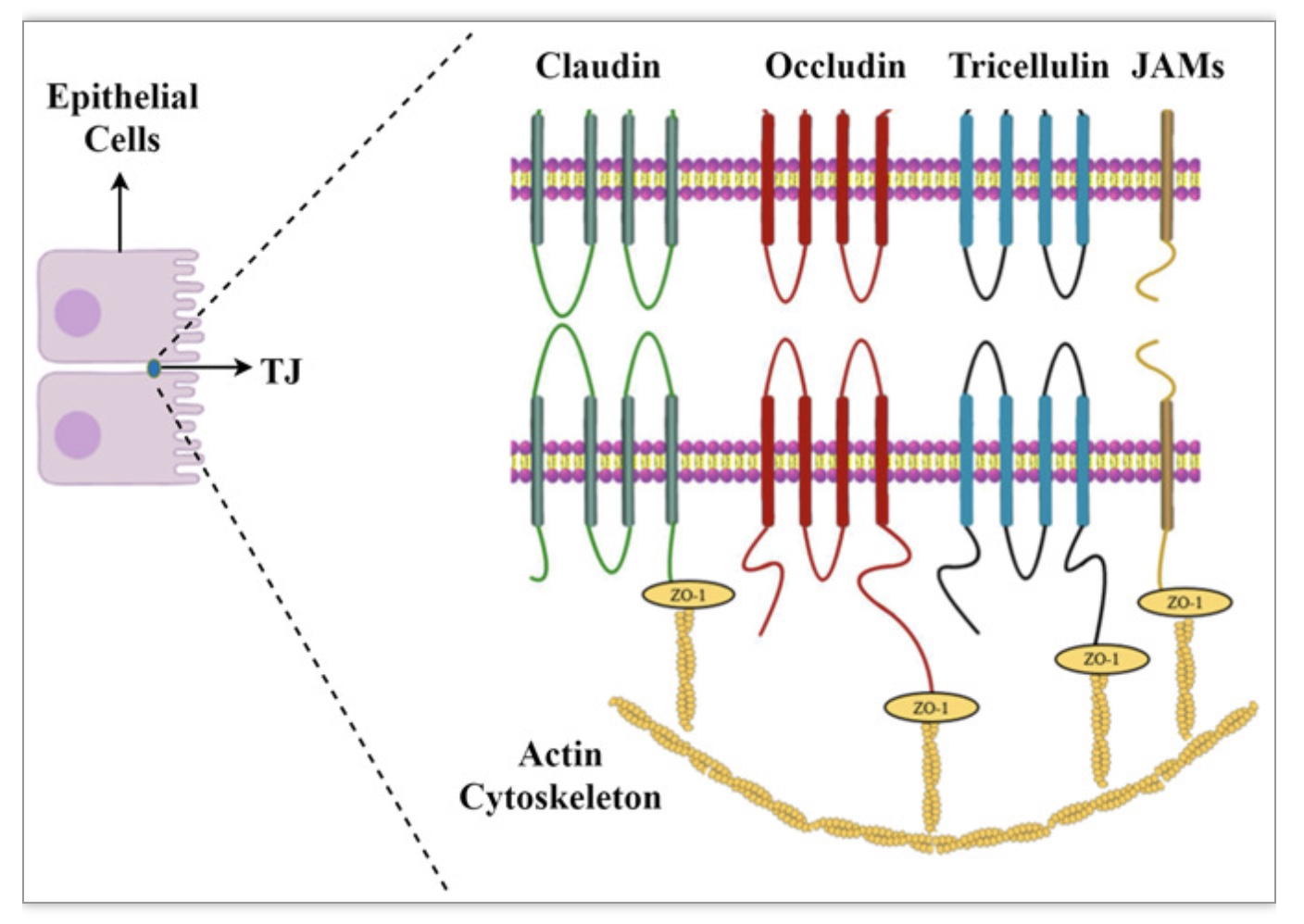
图1. 肾小管上皮细胞中TJs的结构特征
(图源:Wei Wei et al., Journal of cellular physiology, 2023)

图2. TJ蛋白在肾单位的分布特征
(图源:Wei Wei et al., Journal of cellular physiology, 2023)
在缺血再灌注AKI(Ischemia Reperfusion AKI,IR-AKI),TJ蛋白在TECs的表达也发生显著变化。与健康小鼠或正常细胞相比较,ZO-1、occludin、claudin-1、claudin-2、claudin-3、claudin-7、claudin-8、claudin-10、claudin-16、claudin-17和claudin-19的蛋白和基因表达水平发生不同程度的改变,导致肾功能障碍和TJ结构的破坏。同样,在临床的相关研究中也有类似的发现。在肾移植诱导缺血再灌注AKI患者中发现, ZO-1表达明显下调,近端小管TJs完整性受损,导致滤液回漏。以上的数据揭示了TJs异常的表达与IR-AKI密切相关,然而TJs的紊乱与AKI的因果关系尚未明确。
在脓毒症AKI(sepsis-induced AKI,S-AKI)中,ZO-1、occludin、claudin-2、JAM-3的蛋白和mRNA的表达发生不同程度的改变,导致上皮细胞通透性和极性改变、滤液回漏,破坏TJ的完整性,增加粒细胞的粘附。另外,S-AKI通过RhoT1/SMAD-4/JAM-3途径介导TJ破坏,改变上皮通透性。然而,TLR4的敲除,恢复了claudin-2的表达。因此,作者提出了TJs蛋白的改变可能与炎症反应有关。
与其他类型的AKI相似,在肾毒性AKI也与TJ结构的破坏和异常表达有关。肾毒性药物破坏了单层细胞的完整性,导致TJ蛋白的定位发生改变[5]。另外,肾毒性药物可损伤线粒体结构和直接改变TECs的通透性,抑制TJ蛋白的表达,且抑制程度与氧自由基和炎症因子成正相关[6]。
除了上述的AKI类型,其他病因的AKI也可引起TJ改变。如在短期氧化应激暴露诱导的AKI、单侧输尿管阻塞诱导的AKI、烧伤相关的AKI、高氧诱导的新生儿AKI、坏死性肠炎诱导的AKI中,均发现TJ蛋白表达的改变、结构的破坏和功能的异常,揭示了TJ的改变和AKI的发生或许与炎症因子、细胞因子和氧自由基相关。
有研究表明,除了肾脏的TJ损伤外,AKI可能与心、肝、脑、肺等远端器官的TJ损伤有关[7]。在IR-AKI中,脑微血管内皮细胞的TJ蛋白(claudin-5、occludin)和肺泡上皮细胞的TJ蛋白(claudin-4、claudin-18和JAM-1)的表达发生明显改变,这或许是由于AKI期间产生的毒素或促炎因子越过血脑屏障/肺泡-血液屏障导致的远端器官TJ结构的破坏和功能的障碍所致的。这些研究提示了AKI的发生可能与远端器官TJ的损伤密切相关。
综上所述,肾小管上皮细胞TJ结构破坏和TJ蛋白表达异常可能AKI密切相关。越来越多的证据表明TJ的损伤可能参与了AKI的发病机制,但具体的发病机制尚未明确。此外,在AKI的背景下,远端器官(如心、肝、脑和肺)的TJ结构的破坏揭示了肾脏和其他器官之间存在交互作用。各种对肾功能具有保护作用的药物或干预方式,对TJ结构和TJ蛋白表达异常同样具有修复作用。近年来,TJs作为潜在的AKI治疗靶点已成为研究热点。未来需要更多研究深入阐明AKI中TJ蛋白与上下游信号分子之间的动态调控和交互作用机制,为未来的临床应用提供依据。
