
细胞型组合物及其在组织中相对比例是健康的指标。例如,2型糖尿病患者胰腺组织中β细胞团和数量减少 [1, 2],急性髓系白血病(AML)患者表现出与恶性程度相关的细胞类型丰度的变化[3].虽然实验方法如荧光激活细胞分选(FACS)、免疫组化(IHC)和scRNA-seq [4, 5]虽然这些方法可以提供组织中细胞类型比例的最精确测量,但它们面临着高成本、低通量和固体样品中细胞类型分离困难等挑战,使得这些实验方法难以在患者群体规模上应用。为了规避这一问题,科学界利用低成本为感兴趣的组织获取大量rna-seq数据,并配合新兴的机器学习方法领域,通过计算将批量数据反卷积到其组成单元类型组件中[6-9].通常情况下,这些方法使用一组标志基因表征一种细胞类型,从专业知识,将大块组织的平均基因表达反卷积到其组成细胞类型的比例[6, 10]. 后来的方法利用scRNA-seq数据集作为基于细胞类型的基因表达的参考,利用来自所有基因的信息,而不仅仅是标记,提供无标记的反卷积。 [7, 11].其中一些方法不仅在组织中产生细胞型比例,便于临床研究,而且通过将平均基因表达从大量分解为细胞型特异性基因表达,从而实现对基因表达的高分辨率差异分析。 [7, 12]. 后者可以帮助揭示驱动不同组织状态的基因调控程序。

从概念上讲,基因被认为是词汇,细胞被认为是文档,其单词标记(即scRNA-seq读取)是从词汇中取样的,具有CTS主题概率。单链RNA-seq数据假定是从以下数据生成过程生成的。由 m τ{ 1 ,…, M }索引的每个细胞是 K 细胞类型的混合物。细胞型混合物 是从K维Dirichlet分布中采样的,超参数为 特定于细胞。区分GTM-decon与标准LDA的关键假设[14]是用嘈杂的细胞型标签为细胞。标准的主题模型中,主题并不直接与任何已知的概念相关联,需要事后人工检查的基础上,他们得分最高的话来解释他们。引导主题建模方法通过在指导CTS主题混合推理之前,将每个单元格的观察到的单元类型标签以主题的形式加入,从而解决了这个问题。在每个单元格的主题推理过程中,我们通过将单元格类型的主题超参数设置为相对大于其他主题的超参数来引入指导。这样就可以将每个主题锚定到特定的细胞类型,并随后指导对基因的全局主题分布的推断,从而自动确定细胞类型标记基因的优先级。由此产生的主题分布,然后用来推断从散装RNA-seq数据集的相对细胞类型的比例(即,细胞类型反卷积) (图1A所示).作者还将GTM-Decon扩展到推断每个细胞类型的多个主题,以便于捕获由于环境或刺激的变化而产生的不同的细胞状态。最后,作者扩展了GTM-decon使用单细胞或批量转录组数据中可用的表型标签(例如,癌症亚型或癌症分期)作为指南(而不是细胞类型特异性指南)来推断表型特异性(PTS)主题分布,以检测PTS基因表达。 (图一b).进一步扩展到嵌套引导的主题模型,以进行单细胞或成批患者队列数据中CTS差异表达分析 (图 1c ),使用表型标签作为 1 级指南,使用细胞类型标签作为 2 级指南。通过相同的引导主题机制,GTM-decon通过拟合单细胞或批量数据的转录组数据的可能性,更新每个表型下的 CTS 主题分布。
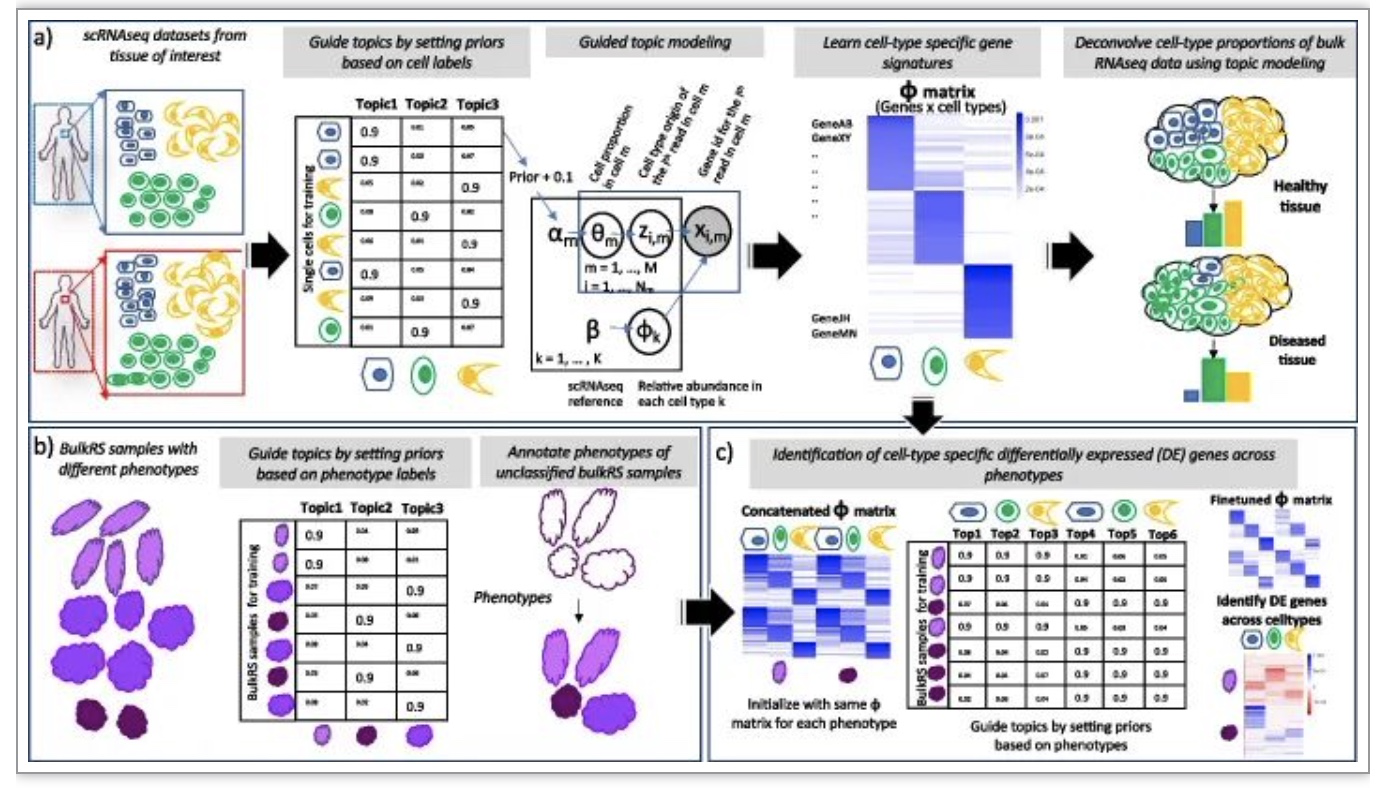
图1:GTM-decon及其应用示意图
(资料来源:斯瓦普纳等人基因组生物, 2023 年)
在5份真实的rna-seq数据中,有3种不同组织类型的已知真实细胞比例。反卷积性能进行了评价,使用斯皮尔曼R和均方根误差(RMSE),通过比较推断的比例对每个样品的地面真实比例的匹配的细胞类型。 结果表明,GTM-decon赋予的标准杆或卓越的性能相比,现有的方法,所有的数据集 (图2a左)。不同细胞类型的反褶积性能 ,根据地面真实比例之间的相关性和预测的比例为每个细胞类型跨样本。总体而言, GT - Decon 授予竞争性能 (图二2c )。GTM-decon通过找到最接近匹配的细胞类型,或通过在细胞类型之间传播信号来适当地表达不确定性,证明了它的鲁棒性。
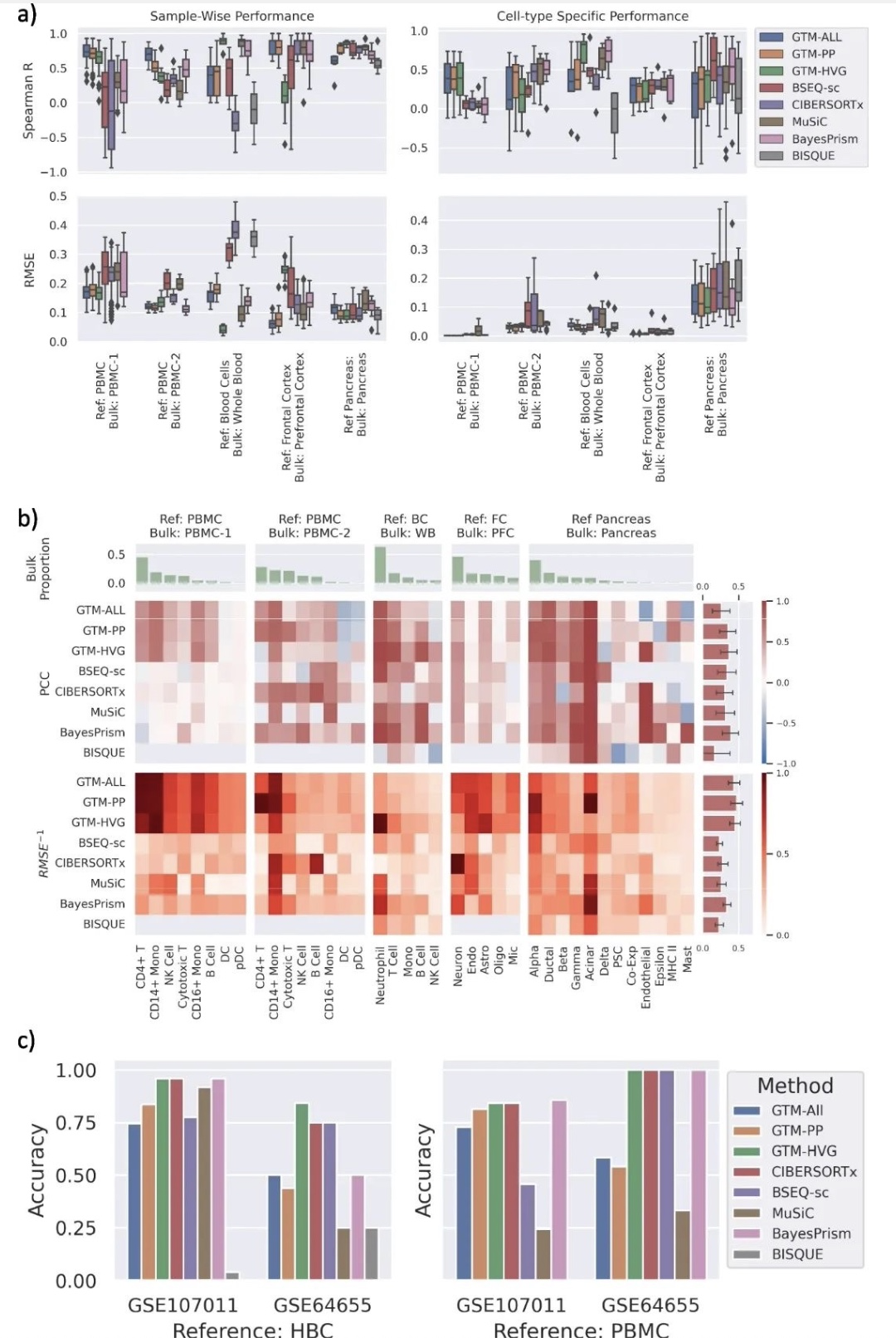
图2:GTM-decon反卷积性能基准测试
(资料来源:斯瓦普纳等人基因组生物, 2023 年)
基于对胰腺和乳房组织进行的实验,作者证明了GTM-decon从scRNA-seq参考中自动学习CTS基因特征,CTS主题下的顶级基因与已知标记基因对已知标记基因信息的细胞类型的富集证明了这一点(图3a).由于GTM-decon推断在每个细胞类型引导的主题下的所有基因的分布,它可以被用来不仅量化的贡献,已知的标记基因,但也得分新的标记基因。此外,作者表明,解旋的细胞型比例可以用来区分健康的和患病的rna-seq样本,就像糖尿病患者的情况一样。 (图二第3条c款e项以及胰腺肿瘤和乳腺肿瘤的癌症亚型 (图四).作者还表明,该方法也可以用来解剖异质性肿瘤微环境。方法确定了两个胰腺肿瘤的不同起源外分泌:导管和腺泡源性胰腺癌(PAAD),胰腺神经内分泌肿瘤(PNET) (图4b),以及富集BRCA亚型的亚型特异性细胞。此外,作者还试图确定大量RNA-seq中可能存在的新的细胞类型或通路,这些在参考图谱中没有出现。这是通过在稀疏的大量RNA-seq数据上运行非引导主题模型(即标准LDA)来检测从头开始大容量RNA-seq(bulkRS)主题.作者还估计了细胞类型比例的变化或大容量的主题是生存时间的指示。为此,使用 Cox 回归来回归患者自癌症诊断以来的生存天数,根据他们推断的细胞类型比例以及从头开始BulkRS主题总体而言,考克斯回归模型与偏倚项(调整后的 p 值= 9 )相比具有统计学意义10-5 基于似然比检验)。为了探讨个体细胞类型比例对生存率的边缘效应,作者基于K-means聚类将患者分为两组进行Kaplan-Meier分析 (图4c).他们观察到,在细胞类型中,内分泌细胞类型表现出显著的风险比,预测了良好的生存结果( p 值=0.0091;log-rank检验) (图4d) 这得到了文献的支持,因为PNET大多是良性的。然而,2型导管细胞与预后差有关,这是因为胰腺癌具有侵袭性(p-值=0.03;log-秩检验)。另一方面,主题1和主题5表明它们的生存能力较差,尽管它们的作用不明确,因为它们不与任何cts主题聚在一起。(图4b).
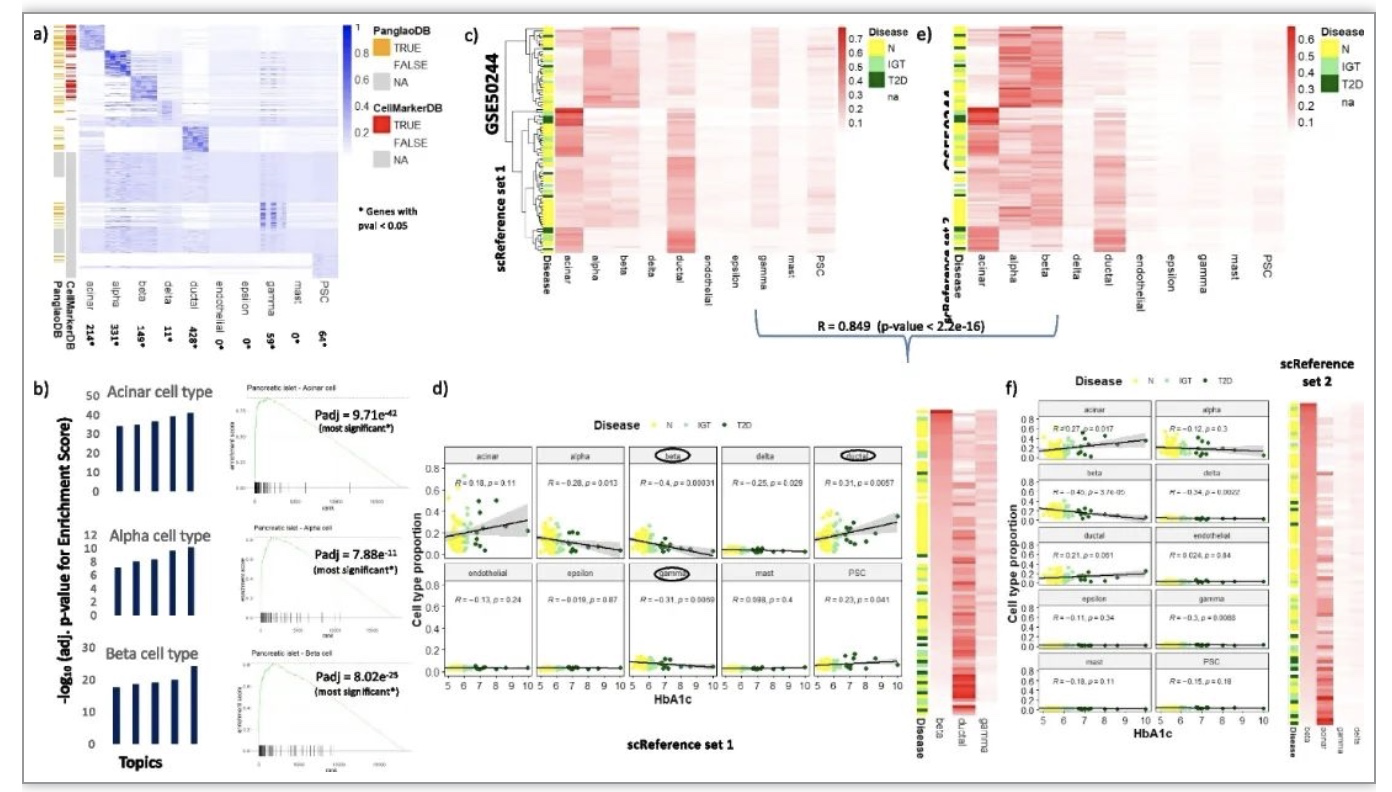
图3:胰腺组织细胞类型特异性主题推断和去卷积
(资料来源:Swapna等,基因组生物学, 2023 )
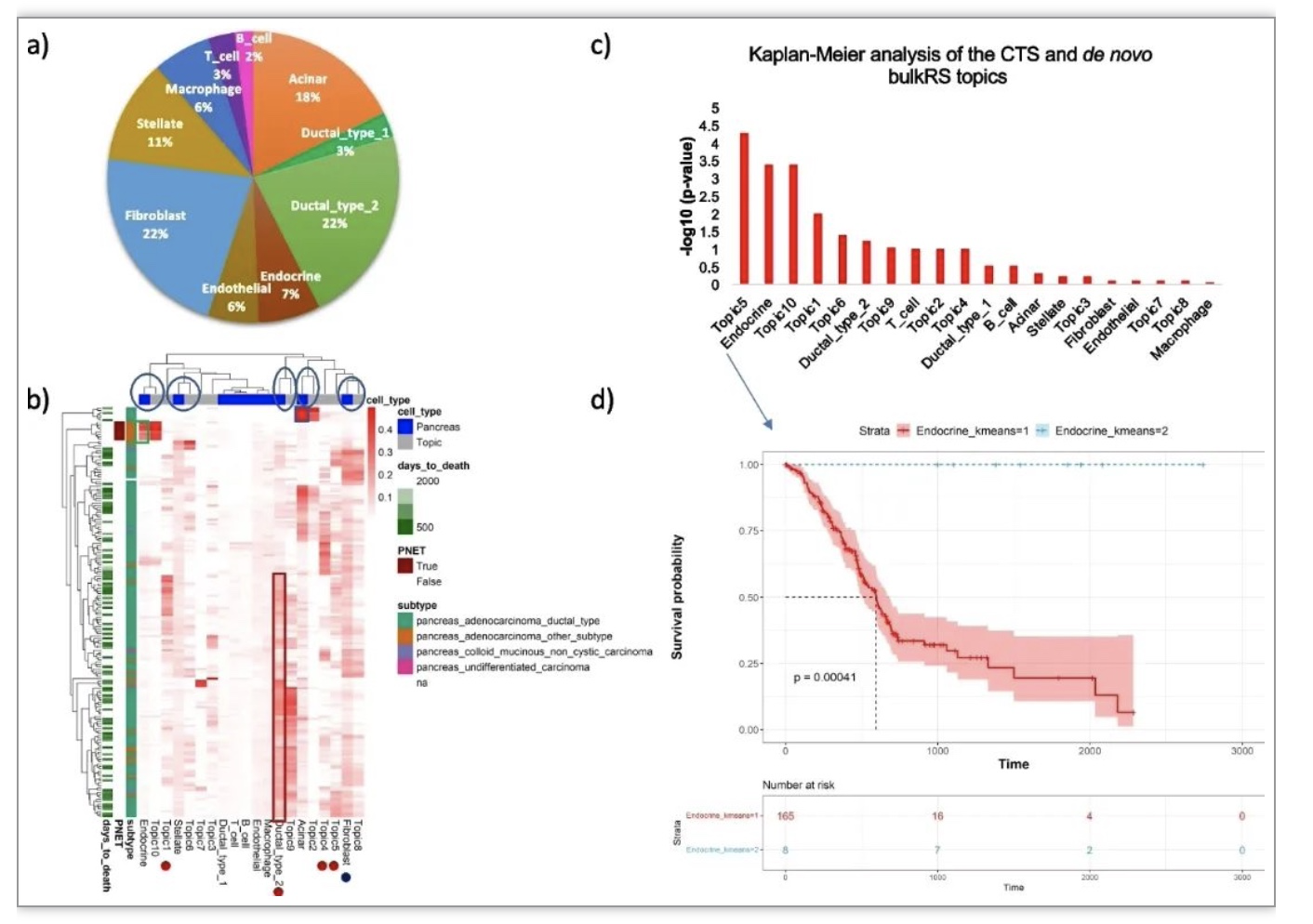
图4所示:胰腺癌大样本RNA-seq的去卷积
(资料来源:Swapna等,基因组生物学, 2023 )
提交人进一步扩展GTM-decon,使用患者表型标签作为指导,而不是细胞类型.与传统的差异分析方法相比,表型引导的GTM-decon提供了一种不同的方法来研究支持感兴趣的表型的基因签名和分子途径。(图一乙)为了在批量数据的上下文中利用scRNA-seq参考数据,作者将这个框架扩展到一个通过从大量RNA-seq数据中为患者的每个表型状态(例如,不同的BRCA亚型)微调一组专用的CTS主题分布,构建嵌套引导的主题模型. 这种方法不仅能够学习细胞类型分布的表型内变化,而且能够学习基因表达表型间变化,从而使作者能够从大量 RNA 序列数据中识别特定表型的 CTS DE 基因(图表 5 )。作者进一步扩展了这种方法 使用单细胞乳腺癌RNA-seq数据的表型和细胞型指南,以癌症亚型为表型指南。模型学习使用表型指南从Sc RNA - seq 数据也导致从大量TCGA-BRCA数据中准确地进行癌症亚型的反褶积以及从单细胞数据本身识别表型特异性 CTS DE 基因表达。
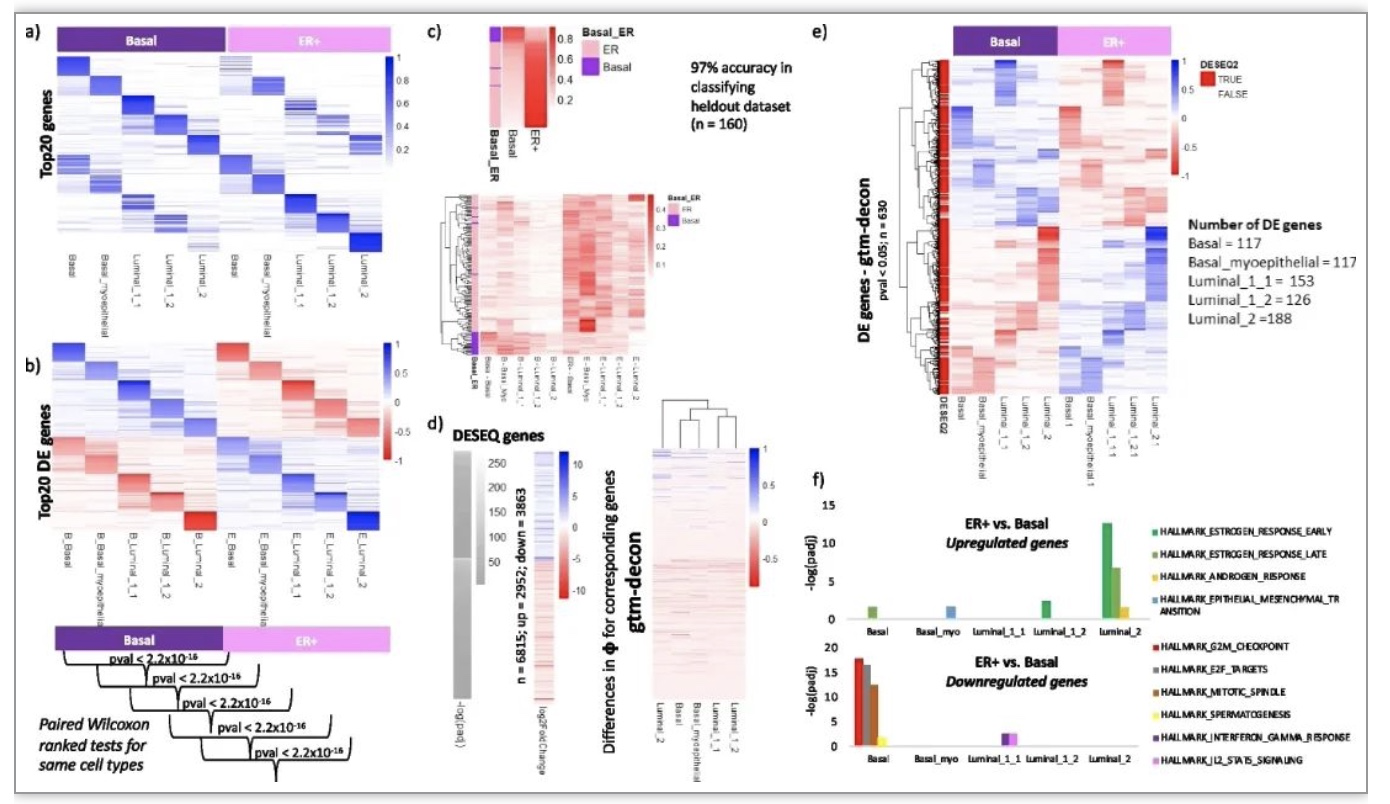
图5所示:从大量RNA-seq数据中鉴定基础亚型与 ER +亚型细胞类型特异性差异表达基因。
结论和讨论、灵感&期待
总之,GTM-Decon是一种有效的无标记反褶积方法,它充分利用了单细胞RNA-seq参考数据进行细胞型反褶积。它提供(1)通过直接模拟大的单细胞RNA-seq参考数据而不使用已知的标记基因来自动推断可解释的CTS主题分布(2)通过推断锚定在相同细胞类型的多个主题来鉴定亚CTS基因表达分布,通过利用表型状态和细胞类型标签信息的扩展嵌套引导主题设计,直接从大量或单细胞样品中检测CTS DE基因。胰腺和乳腺数据集的综合实验演示了所有3个贡献的实用性。像所有其他反卷积方法一样,GTM-decon本身无法从大量RNA-seq数据集中识别新的细胞类型(即,在参考scRNA中不存在的细胞类型)。-seq数据上训练GTM-decon在atlas水平的scRNA-seq数据(如人类细胞景观)上训练GTM-decon[17],它全面覆盖了初级组织中的大多数细胞类型,可以用来规避这个问题。此外,GTM-decon可以利用其他单细胞组学数据,如scATAC-seq作为参考,并对批量样品以及多组学样品中的等效组学进行反卷积。另一个未来的发展方向是利用细胞类型之间的关系,这可能会更好地捕捉潜在的表型状态的主题。
