多组转录因子 (TF) 汇集了多个 CRE,这些 CRE 在基因组上的线性距离可达Mb,以启动和维持其靶基因的表达[1]。因此开发能够分析同一细胞中基因调控多个方面的新方法,包括基因表达、染色质构象、DNA 甲基化、染色质可及性和遗传变异,仍然是功能基因组学领域的长期目标和活跃研究领域。
该研究开发了一种名为NOMe-HiC的多组学研究方法,可在一次检测中同时捕获同一测序片段 DNA 分子的单核苷酸多态性、DNA 甲基化、染色质可及性(GpC 甲基转移酶足迹)和染色体构象变化以及转录组表达。

由于 DNA 三维结构的存在,线性距离较远的CRE可以通过染色质折叠靠近[2],使得空间距离相近的DNA也可能表现出协调的 GpC 甲基转移酶足迹。然而,现有方法不能提供基因组的空间信息来判断染色质可及性的改变是由于转录因子还是远端顺式作用因子的作用[3-5]。
此外,最近的研究表明,杂合单核苷酸多态性 (SNP) 及其相关的 CpG 甲基化或者GpC甲基转移酶足迹同样影响局部 CRE 的活性[6-8]。然而,目前对远程等位基因特异性 CRE 活动的理解仍然受到计算方法和实验技术的限制。染色体构象捕获 (3C) 衍生技术,例如in situ Hi-C[9],空间邻近信息是通过限制酶消化和近端基因组片段的连接来捕获的。连接后,核小体和转录因子仍然与细胞核内的 DNA 交联,并能够被外源性 GpC 甲基转移酶标记,通过后续的亚硫酸氢盐测序检测(WGBS)。因此,研究者基于NOMe-seq[6-8]和 Methyl-HiC[10]技术,进一步开发了 NOMe-HiC,以联合分析来自同一DNA测序片段分子的多组学,以及同一分析中的转录组(图1)。
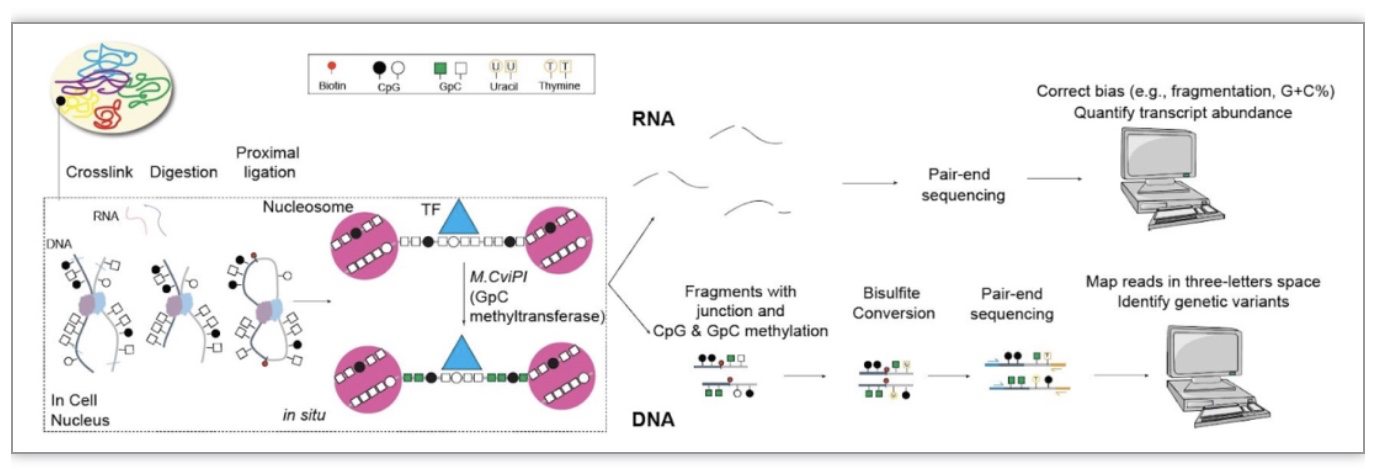
图1 NOMe-HiC技术流程
(图源: Fu, et al., Genome biology, 2023)
为了证明该方法的可靠性,研究者将 3D 基因组,DNA甲基化, GpC甲基转移酶足迹,转录组以及GM2878细胞系的SNP测序结果与相同细胞系中的in situ Hi-C,全基因组亚硫酸氢盐测序 (WGBS),NOMe-seq,ChIP-seq,转录组和深度全基因组测序结果进行了比较,均发现了结果的高度一致性(图2)。
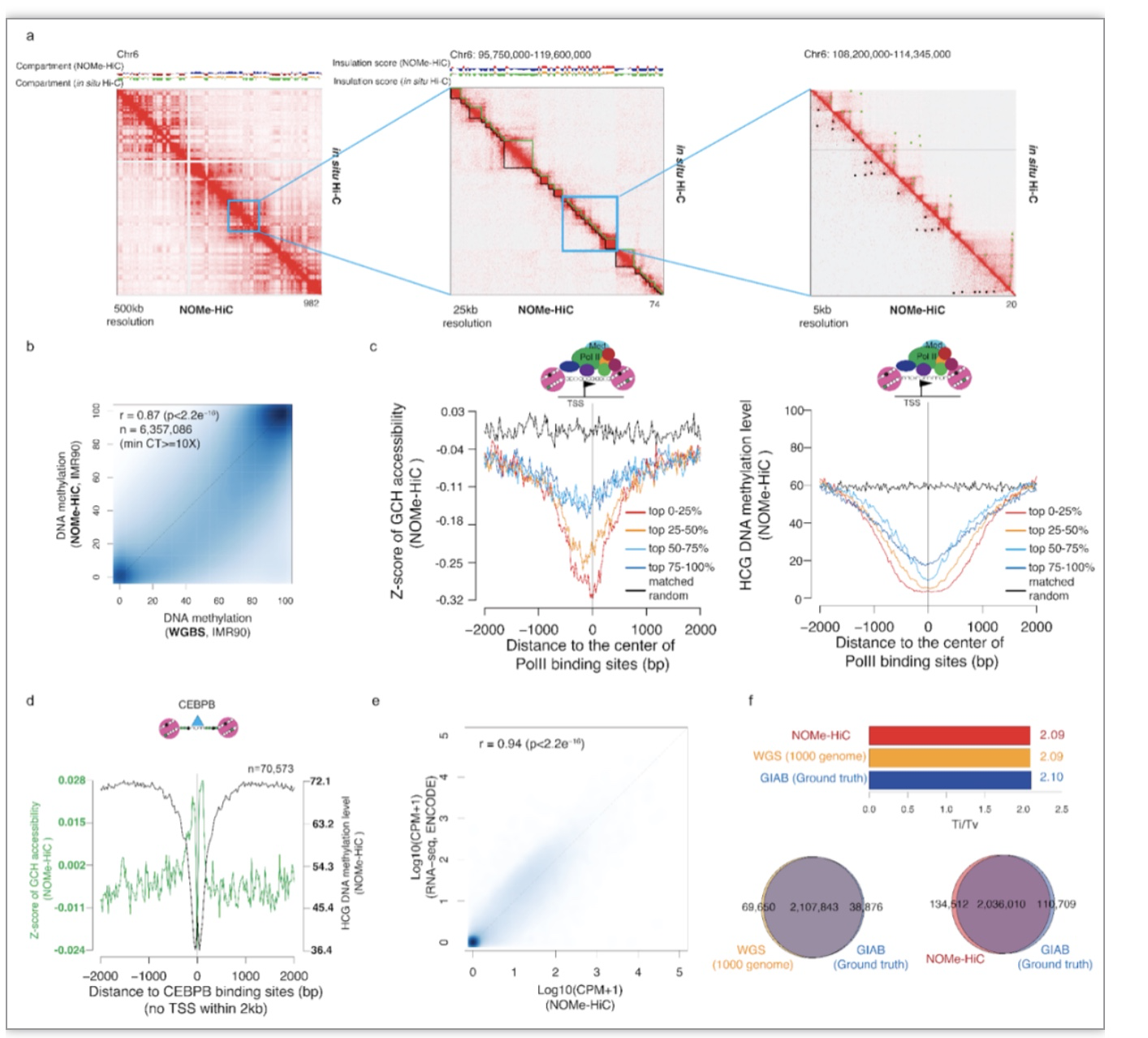
图2 NOMe-HiC数据与现有多组学技术的数据对比
(图源: Fu, et al., Genome biology, 2023)
研究者接下来分析了从分离的染色质环锚中的 NOMe-HiC 读数获得的 GCH 甲基转移酶足迹。结果显示,空间临近的 long-range DNA 分子的染色质可及性的相关性与从local区域的不同 DNA 分子染色质可及性的相关性高度相似,这与我们之前对小鼠胚胎干细胞 (mESC) 的Methyl-HiC 研究一致[10]。然而,与我们之前在 mESCs 中的研究不同,此处 DNA 甲基化的long-range协调性明显低于从人类细胞IMR-90和GM12878,这表明long-range DNA 的内源性甲基化协调性可能在不同物种和不同细胞类型间存在差异。
研究者进一步利用 NOMe-HiC 来分析远程等位基因特异性 GCH 甲基转移酶足迹,并确定了三组等位基因特异性足迹位点。为了表征这些远距离等位基因特异性足迹的功能,研究者分析了已知 TF 结合位点上在锚点的富集情况。第1 组显示两个锚点处 CTCF 结合位点的富集度最高(图 3b),表明 SNP 锚点处的遗传变异对 CTCF 结合的破坏可能影响基因组的loop环结构,进而影响空间邻近的非 SNP 锚点的DNA蛋白结合能力。第 2 组在 SNP 端上表现出最高的 CTCF 和 PolII 结合位点富集,但在非 SNP 锚点上表现出不同 TF 的富集,这些非SNP锚点通常与远端增强子区域结合(图3b)。第 3 组显示出最高的TF 结合位点的缺失(图 3b)。
全基因组关联研究 (GWAS) 将常见的复杂疾病与数十万种遗传变异相关联。在全基因组关联研究(GWAS) 中,与风险或非风险等位基因相关的局部不平衡的染色质可及性经常被用于为同一连锁不平衡块中的潜在的因果SNP提供优势。第 3 组远程等位基因特异性足迹表明,风险和非风险等位基因的转换不需要影响局部染色质可及性,而是影响空间邻近的远距离 CRE 的活动(图3)。实际上,我们观察到 GM12878(B 类淋巴母细胞)中第 3 组的 SNP 锚与四个连锁不平衡区域重叠,这四个连锁不平衡区域与炎症性肠病 (IBD) 显着相关[11-12](图4)。NOMe-HiC提供了一种不需要多个样本的染色质甲基化和可及性QTL作图的替代方法,可以在同一流程中将SNP 与空间邻近的远端CRE 活动的等位基因不平衡联系起来。
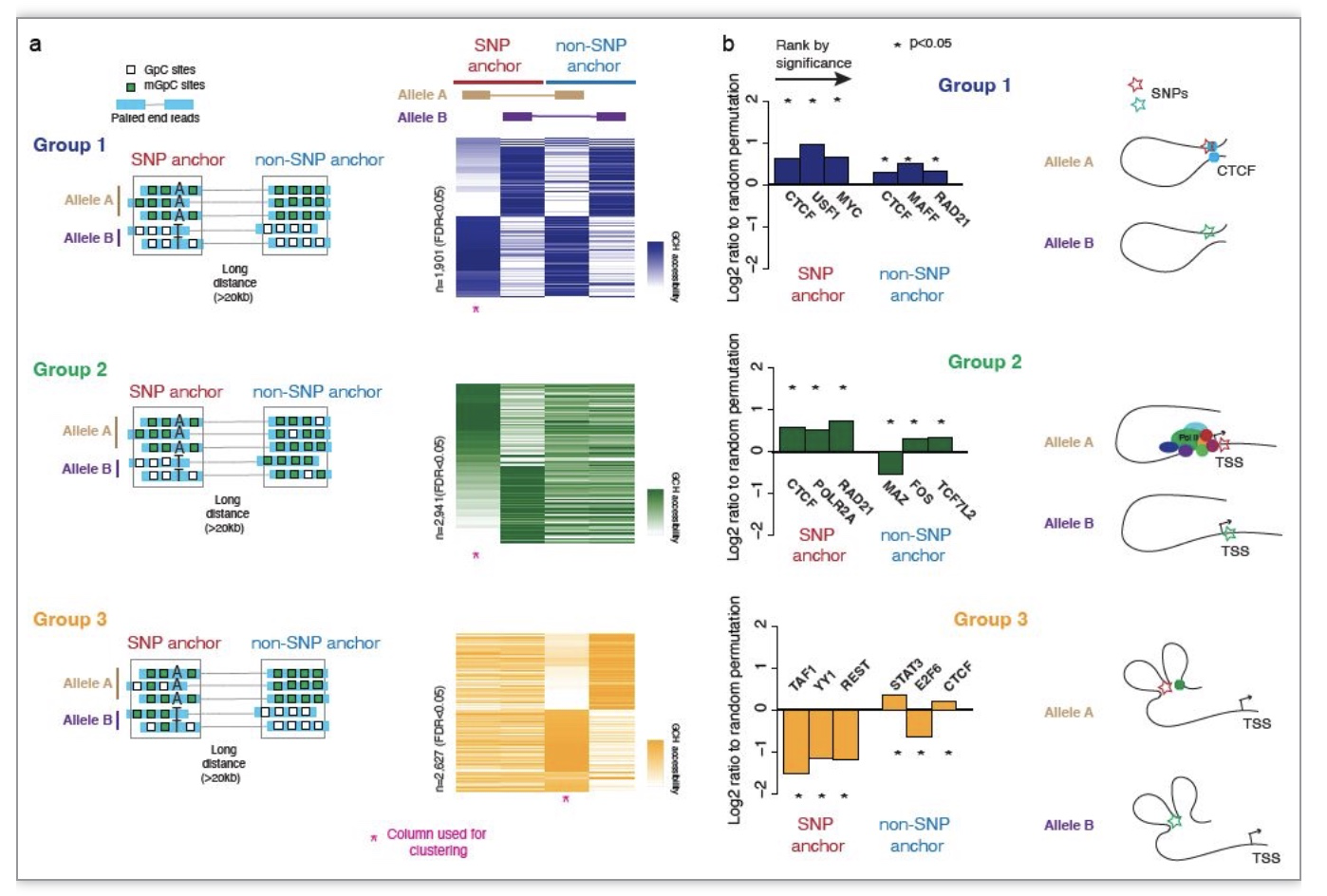
图3 NOMe-HiC揭示long-range等位基因特异性的GCH甲基转移酶足迹
(图源: Fu, et al., Genome biology, 2023)
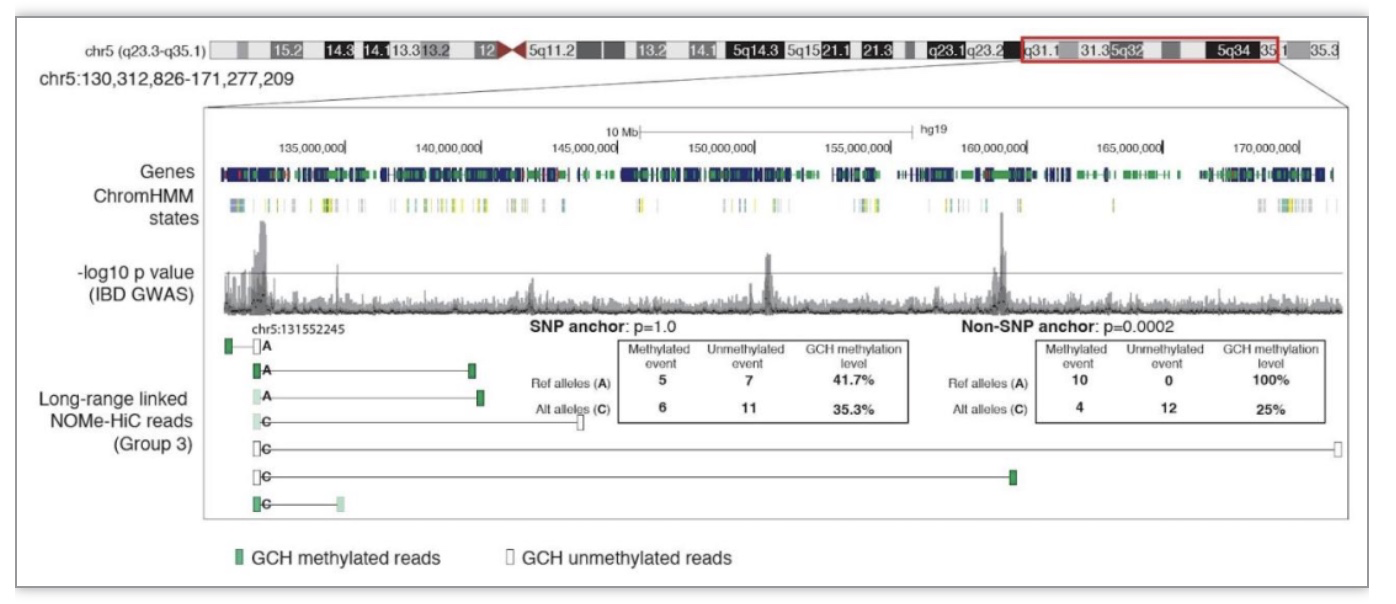
图4 如何使用远程等位基因特异性 GCH 甲基转移酶足迹来注释GWAS 变体的等位基因失衡活动的示例
(图源: Fu, et al., Genome biology, 2023)
综上所述,NOMe-HiC能够用于分析GWAS研究中发现的DNA多态性位点对于没有等位基因失衡的CRE的作用。然而,本研究仍存在一定局限性,比如NOMe-HiC中3D genome结果的分辨率为20kb, 仍不能反映成对CRE的GpC甲基转移酶足迹;亚硫酸氢盐处理对DNA的严重损伤造成大量基因组信息的丢失;二代测序读长的限制使得在测序结果片段钟许多线性距离非常远的CREs不能被捕捉到。未来,将NOMe-HiC与三代长链测序技术和更温和的DNA甲基化检测方法结合,将有效提高NOMe-HiC的对于空间临近CRE的表观遗传状态的分析能力。

