
在慢性肝病发展为肝硬化的过程中,肝细胞死亡引起肝细胞减少从而导致肝功能异常、失代偿、肝衰竭,甚至死亡。以往对于肝细胞死亡的研究主要关注细胞凋亡和细胞坏死两种方式。在致病因素长期作用下,临床肝硬化患者肝脏有不可自限的炎症反应和肝细胞消亡。细胞凋亡不引起组织炎症,细胞坏死多出现在急性肝损伤中,这提示肝硬化肝脏中可能存在新的细胞死亡形式。
该研究发现了干扰素刺激因子(STING)通过NLRP3炎性小体诱导肝细胞焦亡促进肝脏纤维化,并揭示了STING通过WDR5/DOT1L促进Nlrp3启动子区组蛋白甲基化增加IRF3募集,从而增加NLRP3表达的分子机制。

首先,通过对CCl4诱导的肝纤维化小鼠肝脏进行了RNA测序,发现STING和NLRP3炎性小体信号通路在肝纤维化时被激活,这两条通路的激活也在人和小鼠肝硬化组织中得到了验证。肝细胞STING在小鼠和人肝硬化组织中显著增加,而敲除Sting基因以及使用STING抑制剂C-176均可缓解CCl4诱导的肝纤维化。由于STING和NLRP3信号通路在肝硬化时都被激活,且敲除Sting和STING抑制剂C-176均可以显著抑制肝细胞NLRP3表达和肝细胞焦亡,提示STING可以通过活化NLRP3炎性小体诱导肝细胞焦亡。因此,该研究的核心创新之一是验证STING对NLRP3炎性小体信号通路和肝细胞焦亡的表观调控。
在表观调控机制研究中,研究使用TNFa刺激Sting过表达的肝细胞,建立STING依赖的NLRP3高表达肝细胞体系,然后使用组蛋白修饰酶小分子抑制剂库进行干预,最终发现H3K4特异性组蛋白甲基转移酶WDR5和H3K79甲基转移酶DOT1L抑制剂可以显著降低Sting过表达介导的Nlrp3上调(图1A-E),这个过程依赖于STING下游IRF3的激活,而不依赖于NFkB。接着,研究通过免疫共沉淀(CoIP)证实了WDR5、DOT1L和STING关键下游转录因子p-IRF3结合(图1F-G)。此外,通过染色质免疫共沉淀(ChIP)实验表明,TNFa和STING激动剂DMXAA刺激肝细胞后,p-IRF3,H3K4me2以及H3K79me3在Nlrp3启动子区域结合增加(图1H-J),说明STING通过WDR5/DOT1L促进Nlrp3启动子区组蛋白甲基化,从而募集IRF3增加Nlrp3转录。
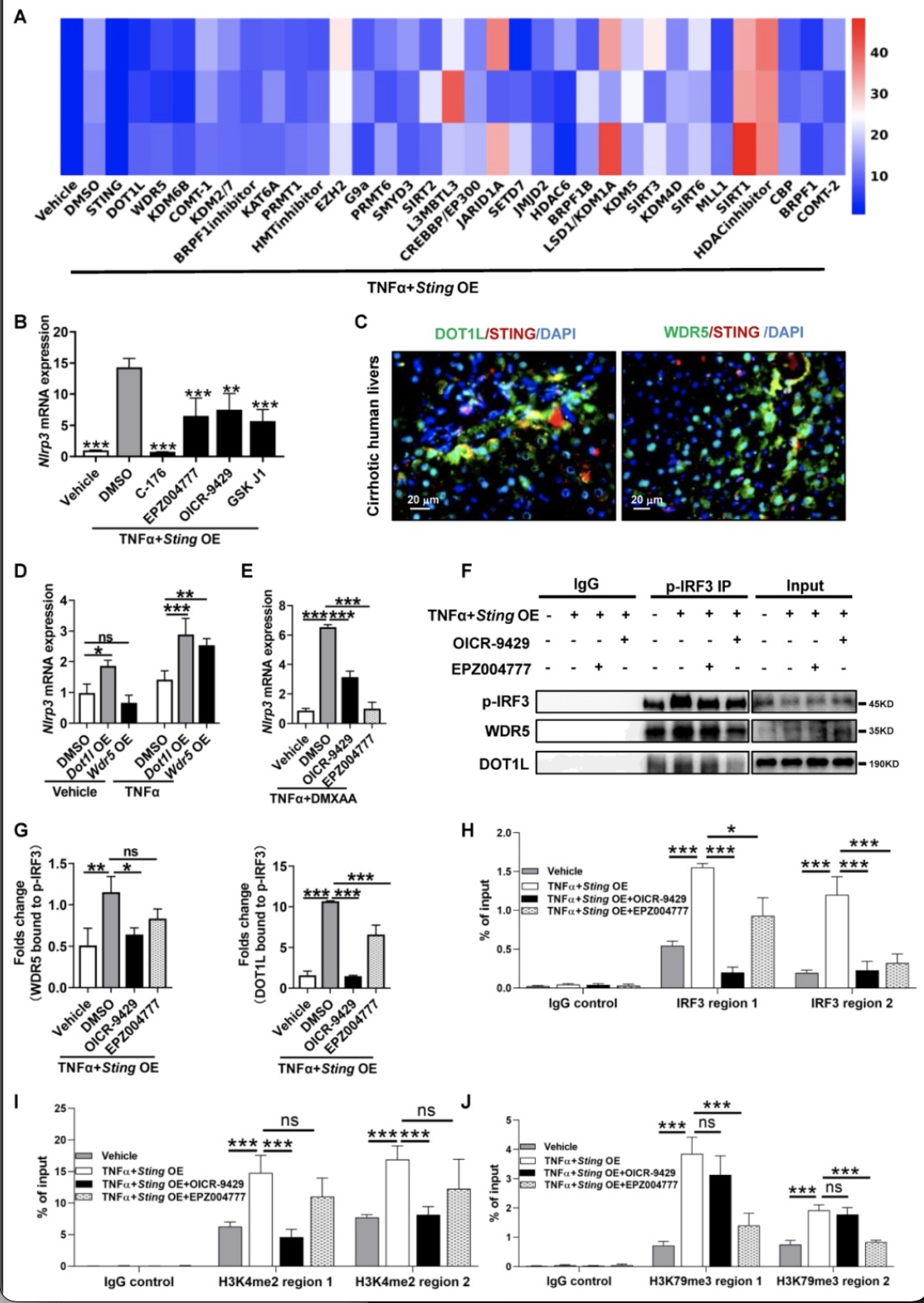
图1 STING通过WDR5/DOT1L促进Nlrp3启动子区组蛋白甲基化,招募IRF3促进Nlrp3转录
(图源:Xiao, et al., Redox Biology, 2023)
在阐明了STING对NLRP3的表观调控机制后,该研究进一步探索了肝细胞NLRP3和肝细胞焦亡对肝脏纤维化的影响。作者发现Nlrp3肝细胞特异性敲除可以显著缓解肝脏纤维化和肝脏炎症浸润,改善肝脏功能。为突出NLRP3这个靶点的临床转化价值,论文发现使用NLRP3抑制剂MCC950可显著改善CCl4诱导的小鼠肝纤维化。此外,通过敲除NLRP3下游诱导肝细胞焦亡的关键蛋白GSDMD,也可以通过减少肝细胞焦亡缓解TAA和BDL诱导的肝纤维化。论文第二个核心创新是通过临床前研究发现STING、NLRP3、GSDMD是治疗肝硬化的新靶点。
虽然有肝细胞焦亡促进NASH相关肝纤维化的研究,但肝细胞焦亡促进肝纤维化尚不清楚。该研究通过分析前期小鼠肝纤维化组织和焦亡肝细胞RNA-seq数据,发现肝细胞代谢相关信号通路紊乱、ROS相关通路激活。因此该研究进一步验证了STING敲除和Nlrp3肝细胞特异性敲除可以通过缓解肝脏代谢重编程和肝细胞ROS产生,从而抑制肝星状细胞活化,最终缓解肝纤维化。论文第三个核心创新是从ROS和代谢重编程的角度阐明了肝细胞焦亡促进肝纤维化的机制。
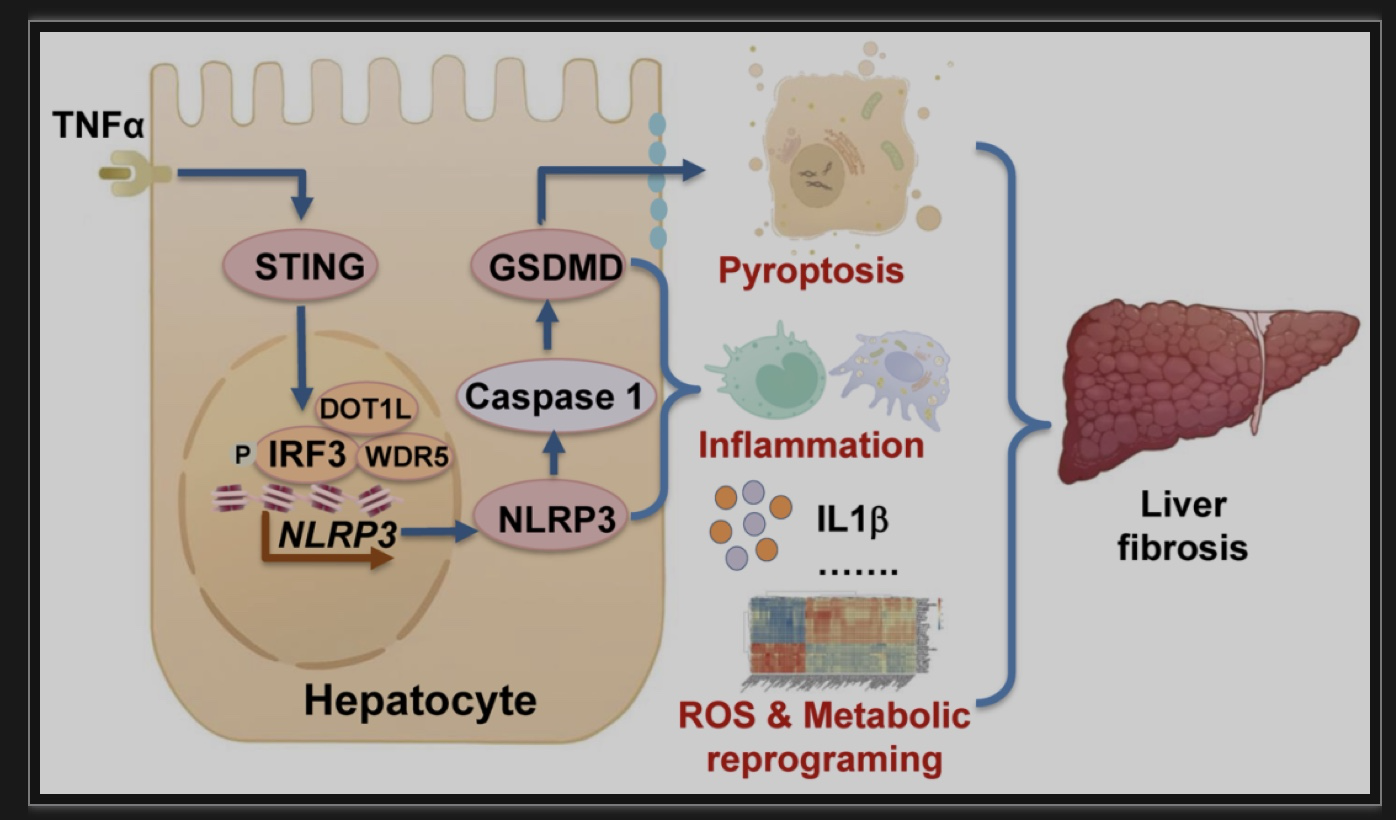
图2 研究机制图
(图源:Xiao, et al., Redox Biology, 2023)
