
细胞增殖依赖于乙酰辅酶A,其是脂质合成和乙酰化反应的关键前体物质,在生物膜形成,能量储存和蛋白修饰过程均发挥着重要的作用[1]。在应激条件下,线粒体,过氧化物酶体和细胞质醋酸盐的活化都共同维持着乙酰辅酶A的稳态。线粒体ATP-柠檬酸裂合酶(ATP-citrate lyase, ACLY),细胞质乙酰辅酶A合成酶(Acetyl-CoA synthetase, ACSS2)和过氧化物酶体生物发生因子5(Peroxisomal biogenesis factor 5, PEX5)是乙酰辅酶A形成的关键酶。有研究报道,ACLY和ACSS2的高表达和肿瘤的发生高度相关[2, 3],药理抑制和基因敲除被证明可减少脂质生成和细胞增殖;另一方面,细胞中营养供给和线粒体代谢有时在肿瘤微环境中受到损害,其他代谢通路通常无法完全代偿[4]。寻找一种乙酰辅酶A生成的替代途径对于理解癌细胞的增殖和抵抗机制尤为重要。虽然正常和肿瘤组织都能从食物中获取脂质并从头合成以维持细胞增殖[5],然而这些代谢过程涉及的通路和细胞器在是如何协调以供给脂质生成的还不完全清楚。
该研究利用质谱,稳定同位素示踪,和同位素谱分析(ISA)分析当癌细胞中的乙酰辅酶A的合成过程被系统性破坏时的代谢重塑过程。研究者利用13C同位素示踪细胞,发现在多种细胞中缺失ACLY脂肪酸合成减少,细胞对于细胞外脂质或醋酸盐的摄取增加。同时敲除ACLY和ACSS2后细胞生长受限,但并未完全阻止增殖,表明存在替代途径可以支持乙酰辅酶A的稳态。代谢流示踪和PEX5敲除的实验表明在缺失ACLY的细胞中,过氧化物酶体氧化外源性脂质是脂质生成和组蛋白乙酰化所需乙酰辅酶A的主要来源,强调了细胞器间“Cross-talk”在营养波动时支持细胞存活的作用。

经典的脂质从头合成过程如图1A所示,ACLY和ACSS2对乙酰辅酶A的生成起着关键作用。为了理解在癌细胞中ACLY和ACSS2是如何对脂肪酸合成贡献的,研究者利用CRISPR-Cas9系统构建了A549的敲除细胞系(图1B),发现ACLY缺失后细胞的生长速度明显变慢(图1C),葡萄糖摄取和乳酸盐的外排速率明显降低;谷氨酰胺的摄取和谷氨酸盐的外排速率明显升高,这些结果表明碳代谢过程发生重塑。接下来,研究者利用ISA定量测定了棕榈酸的合成和U-13C6葡萄糖对新生成的乙酰辅酶A的贡献。结果显示,与对照细胞比较,ACLY缺失细胞棕榈酸酯合成大幅减少(图1F)。值得注意的是,仍有20%左右U-13C6葡萄糖来源的乙酰辅酶A(图1G)。
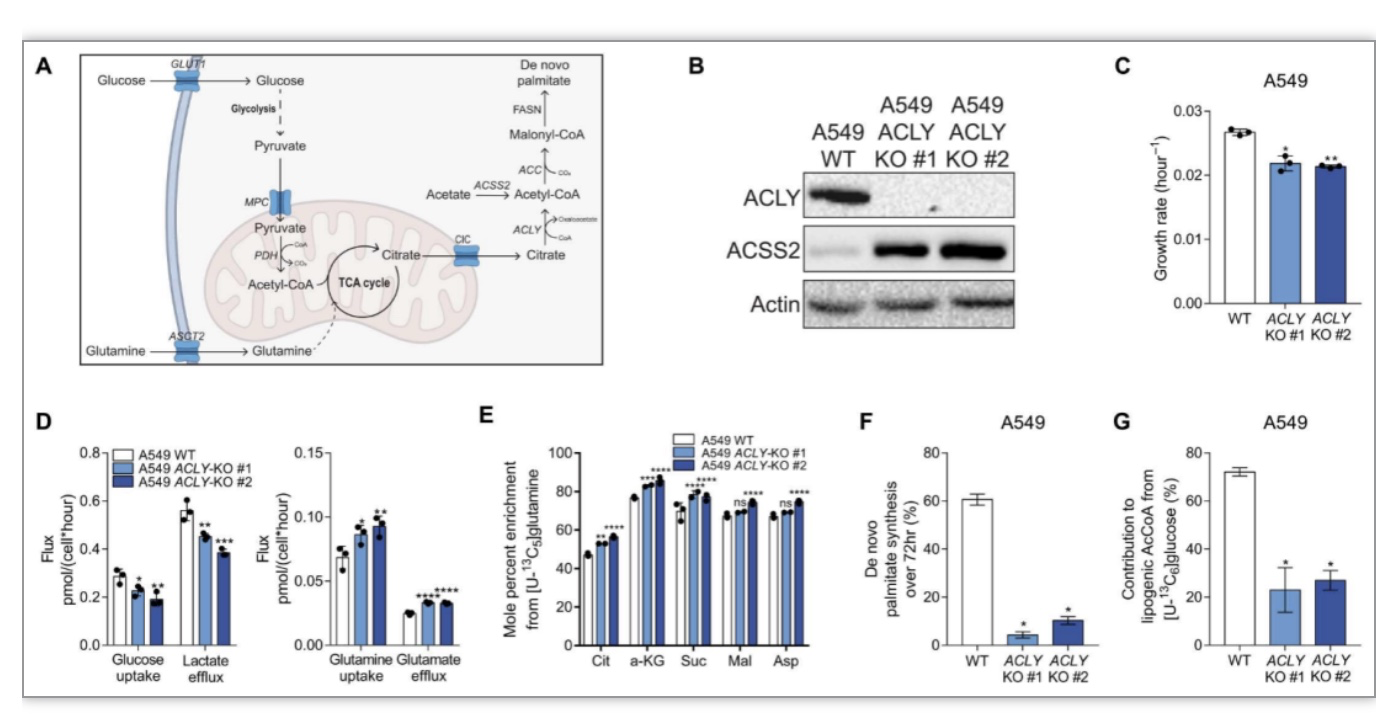
图1 在癌细胞中缺失ACLY重塑碳代谢,棕榈酸合成速率降低
在葡萄糖来源的柠檬酸盐供给受限的情况下,例如缺氧时,醋酸盐可被乙酰辅酶A合成酶“激活” ,由图1B也能够发现,ACLY缺失细胞中ACSS2蛋白表达显著升高(图1B)。作者由此假设:外源提供醋酸盐是否可以回补ACLY的缺失。结果显示,在细胞培养基中添加醋酸盐能够回补缺乏ACLY细胞的棕榈酸的合成(图2A)。不仅如此,醋酸盐也是ACLY敲除细胞的主要脂质生成底物 (图2B)。另外在ACLY缺失的细胞中补充醋酸盐可促进细胞增殖,尤其是在缺乏脂质的情况下(图2C),这表明乙酰辅酶A的供给存在冗余的过程,即当通过ACLY供给乙酰辅酶A的代谢过程被阻断时,通过ACSS2供给过程增强(图2D)。作者也做了ACSS2的敲除细胞系,不出所料,ACSS2的缺失,外源供给的醋酸盐整合进入乙酰辅酶A的减少50%以上(图3A,B),然而棕榈酸的合成和细胞的生长却几乎不受影响(图3C,D)。
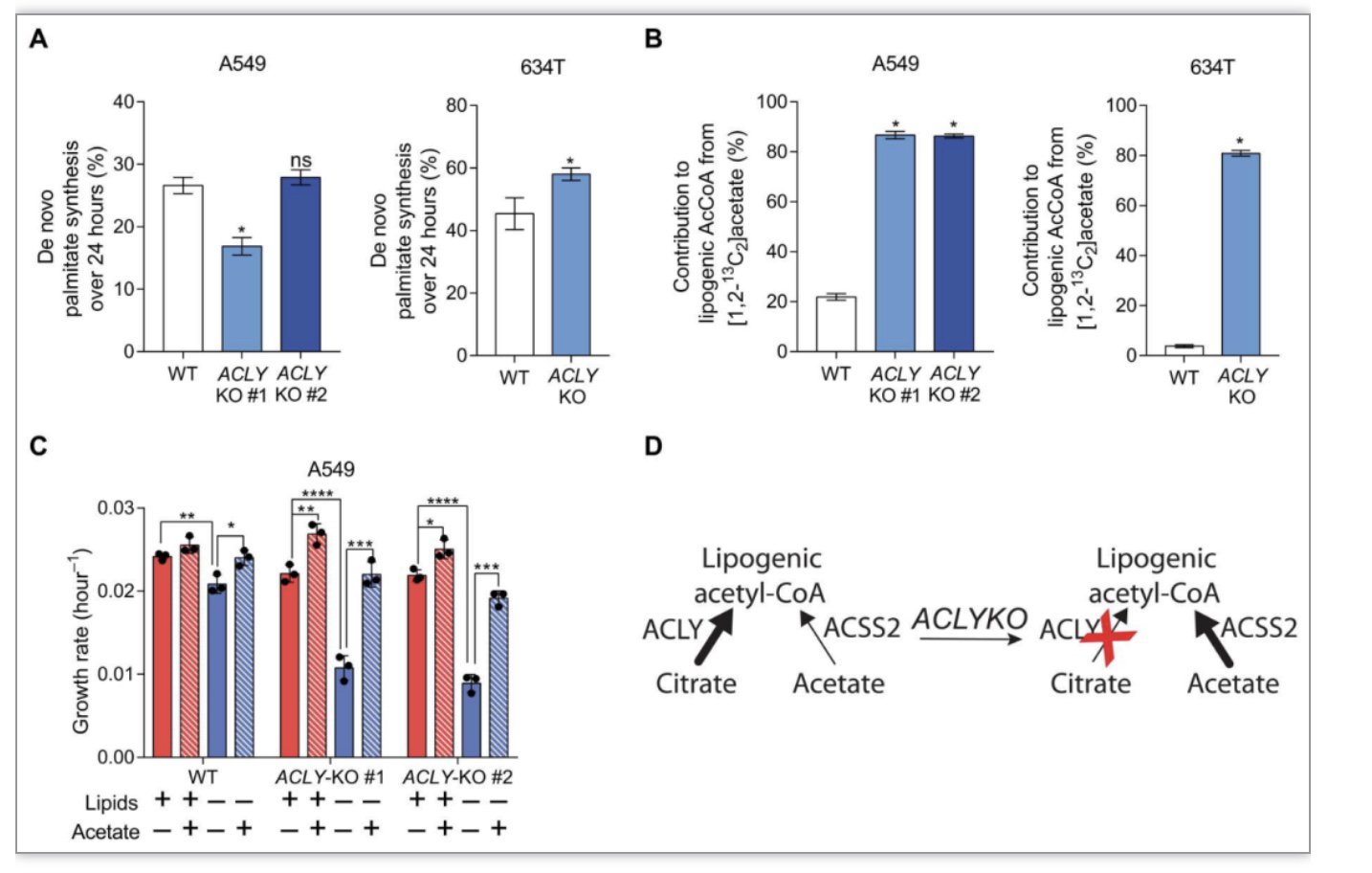
图2 外源添加醋酸盐能够回补ACLY缺失导致的细胞增殖减缓和脂肪酸合成速率下降
接下来,研究者构建了ACLY/ACSS2双敲细胞探索癌细胞是如何响应乙酰辅酶A来源的极端限制的(图4A)。结果显示,相较于野生型细胞,ACLY/ACSS2双敲细胞生长明显受损,即便外源添加醋酸盐也无明显的回补效果 (图4B)。用同位素标记的葡萄糖或醋酸盐示踪新合成的棕榈酸,未观察到同位素的富集 (图4C,D),这些结果证实在双敲细胞中ACLY/ACSS2所在的两条乙酰辅酶A合成的通路均存在缺陷。哺乳动物细胞不能合成多不饱和脂肪酸(Polyunsaturated fatty acid, PUFAs)而需要依靠外源供给。相较于野生型和ACSS2敲除细胞,ACLY单独缺失和ACLY/ACSS2双敲细胞整合PUFAs转化为磷脂酰胆碱的比例明显升高,表明在ACLY缺失的细胞中,外源性脂肪酸对膜脂质的贡献增加(图4E)。尽管有这些脂质代谢的变化,乙酰化组蛋白acH3K27水平在ACLY单独缺失和ACLY/ACSS2双敲细胞间无明显差异(图4G),表明癌细胞有独立于ACLY和ACSS2的通路的其他的来源维系乙酰辅酶A的供给。接下来,作者通过一系列同位素示踪实验排除了ACLY和ACSS2双敲时线粒体其他可能参与乙酰辅酶A生成的关键调控分子,如肉碱乙酰转移酶(Carnitine acetyl-transferase, CRAT), 线粒体丙酮酸载体(Mitochondrial pyruvate carrier (MPC)。
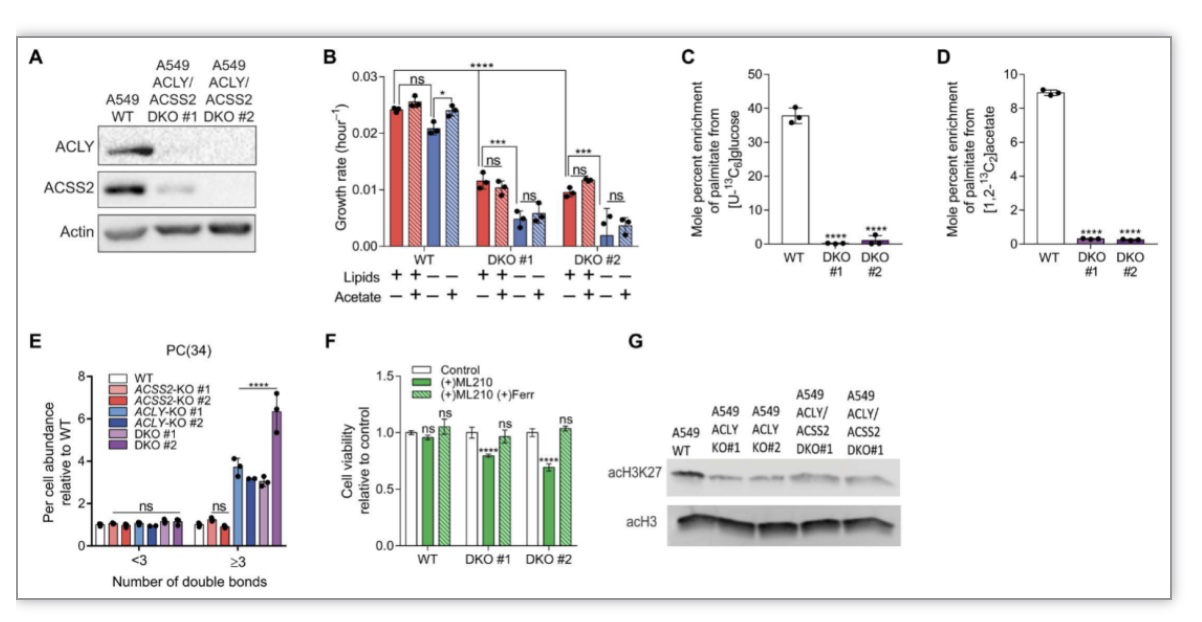
图3 ACLT/ACSS2双敲细胞依赖于细胞外来源的脂质
已知乙酰辅酶A的一个关键来源是过氧化物酶体通过脂肪酸的β-氧化产生,通常是长链脂肪酸(VLCFA)或支链脂肪酸(BCFA)。Izzo等也几乎同时报道过ACLY/ACSS2双敲细胞中过氧化物酶体脂肪酸氧化的相关基因高表达[6]。为了探究过氧化物酶体在癌细胞中是否可以作为一种有效的乙酰辅酶A的来源,研究者构建了野生型和ACLY/ACSS2敲除细胞的脂质丰度谱图,发现大多数敲除细胞的甘油三酯略有减少而磷脂不变(图6A)。然而,脂质的酰基链组成发生了重大变化,即ACLY缺失和ACLY/ACSS2 双敲细胞中包含VLCFAs的甘油三酯和磷脂丰度极低(图6B,C),其可能的一个原因是VLCFA在过氧化物酶体中的氧化增加。
于是研究者将目光转向过氧化物酶体在乙酰辅酶A生成中的作用。PEX5是过氧化物酶体行驶功能的关键分子,过氧化物酶体中的多数酶是借助受体PEX5从胞浆中转运进入的。为了进一步研究过氧化物酶体在支持乙酰辅酶A代谢中的作用,研究者构建了PEX5单敲和ACLY/PEX5双敲细胞系并给与细胞U-13C8辛酸盐处理,发现无论是新合成的乙酰辅酶A还是脂肪酸的合成,在PEX5/ACLY双敲细胞的量中都显著低于单敲细胞(图6D-F),此结果证实了过氧化物酶体支持乙酰辅酶A和脂质合成,即敲除PEX5破坏了ACLY 敲除细胞中过氧化物酶体脂肪酸氧化对乙酰辅酶A生成的贡献(图6G)。为了进一步支持缺失ACLY后细胞中过氧化物酶体氧化增加的观点,研究者利用长链脂肪酸U-13C18油酸示踪细胞。结果显示,相较于野生型细胞,ACLY缺失细胞中从U-13C18油酸而来的溶血磷脂酰胆碱显著增加,表明细胞对外源性脂肪酸氧化供给的依赖性增加(图6H)。当同时敲除PEX5,LPC 16:1的丰度如预期降低(图6H)。
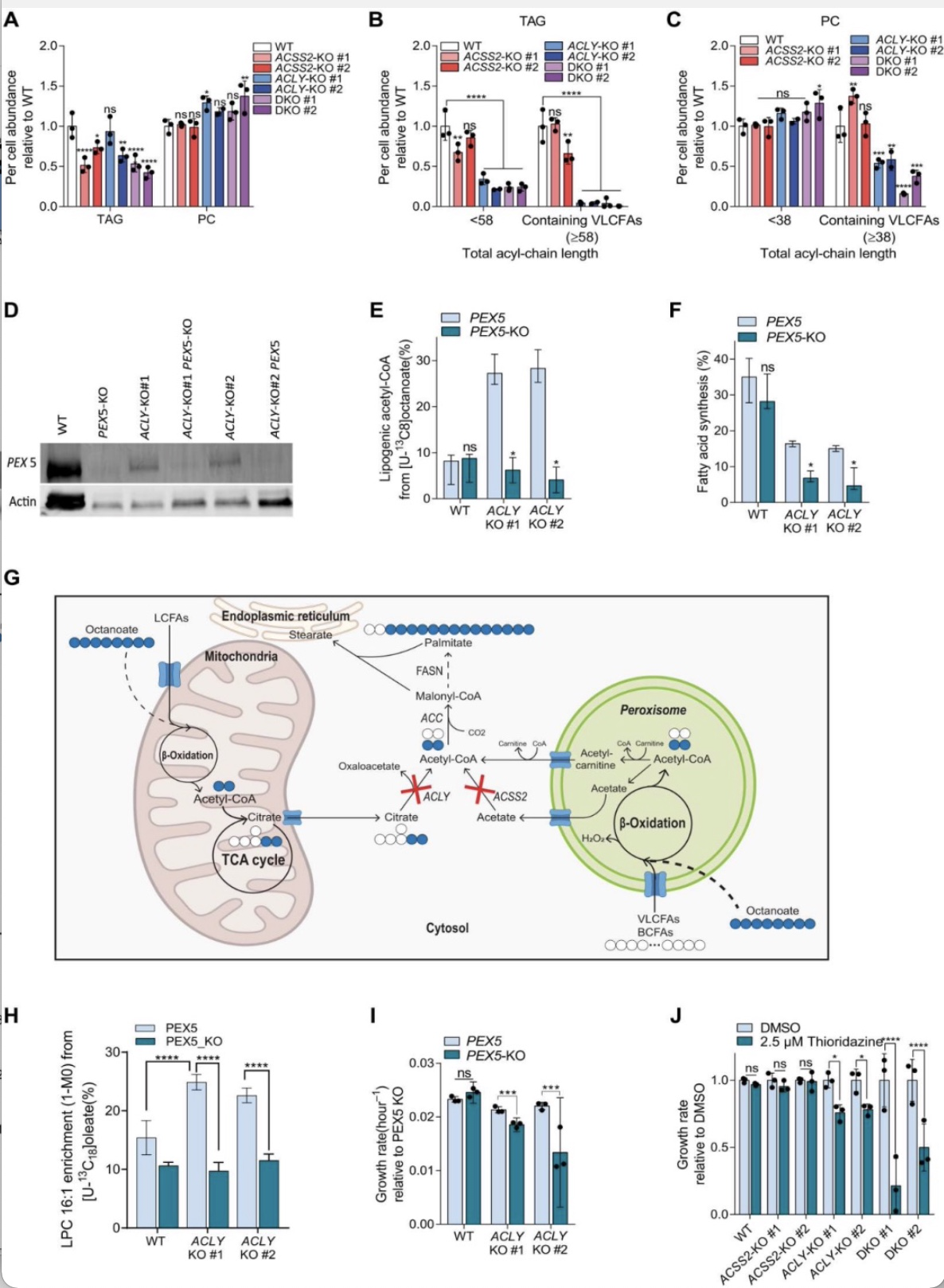
图4 过氧化物酶体β-氧化在ACLY和ACSS2缺失时供给乙酰辅酶A
综上所述,这篇论文首先证明了在细胞中缺失ACLY能够减少脂肪酸合成,并代偿性的增加细胞对细胞外脂质或醋酸盐的摄取,以供给乙酰辅酶A。接着设计双敲ACLY和ACSS2的实验进一步提示存在替代途径可以支持乙酰辅酶A的稳态。最后证明过氧化物酶体氧化外源性脂质是亦是细胞营养波动时乙酰辅酶a的主要来源,强调了细胞器间Cross-talk的作用。文章略显不足的是:缺乏动物的生理数据支持;线粒体和过氧化物酶体均表达CRAT,在贡献乙酰辅酶A上的分配过程上研究的不够清楚。
