
白色念珠菌是一种常见的能与人体共生的真菌,可引起免疫缺陷人群浅表或深部真菌感染。随着免疫抑制剂在器官移植手术中的广泛应用、艾滋病患者人数逐年升高以及人口老龄化等高风险因素越来越多,免疫系统受到抑制或损伤的情况越来越普遍[1]。尽管临床上已有一些抗真菌药物应用于治疗真菌感染,但侵袭性念珠菌病患者的病死率仍然高达46-75%[2]。侵袭性念珠菌感染极高的死亡率,反映出目前的抗真菌治疗受限于抗真菌药物库不足,以及药物毒性和真菌耐药性等问题的困扰。因此,发现新型安全高效的抗真菌药物具有重要临床意义。白色念珠菌在感染过程中会产生多种毒力因子如菌丝、被膜等,这些毒力因子使得白色念珠菌能更好的适应复杂多变的宿主环境,增强其在体内致病能力,在感染中发挥着重要的作用[3]。与常规抗真菌药物相比,毒力抑制剂由于不影响真菌细胞的正常生长,造成的选择压力小,不易产生耐药,因此毒力抑制剂的开发具有很好的临床应用前景。
该研究基于群体感应分子法尼醇和甾醇合成通路的联系,通过高通量筛选得到了白色念珠菌毒力抑制剂H55。机制研究表明H55可以特异性抑制白色念珠菌甾醇合成通路上的甾醇甲基转移酶(Erg6),并揭示了Erg6的功能缺失会影响菌丝形成调控网络,导致菌丝形成等毒力因子的缺陷,该研究为Erg6作为抗毒力靶点进一步开发提供了理论支持。

法尼醇是白色念珠菌自身分泌的一种群体感应分子,可以抑制菌丝形成。法尼醇合成通路是甾醇合成通路的一条侧支,许多甾醇通路合成抑制剂如氟康唑和zaragozic acid都会导致法尼醇合成的增多。为了筛选得到Dpp3高表达的诱导剂,作者首次建立了基于法尼醇合成酶Dpp3绿色荧光标记的毒力抑制剂筛选模型[4],对约4万余个小分子(来源于国家化合物样品库和自有天然小分子库)进行高通量筛选(图1A)。作者对其中诱导荧光强度升高幅度最高的安替比林类衍生物H55(图1B,C)进行了一系列毒力抑制能力检测,结果显示H55在多种菌丝诱导条件下表现出菌丝形成抑制作用,并能抑制对哺乳动物细胞的粘附和被膜形成(图1D-1H),而H55本身无明显抑制真菌增殖作用,对正常细胞半数抑制浓度(IC50)也远高于毒力抑制所需浓度。为了确证H55在体内治疗的应用潜力,作者首先使用两种感染模型对H55进行评估,结果显示H55在小鼠口腔粘膜感染模型和系统性模型中均表现出了较好的治疗效果(图2)。H55低毒和高效抑制毒力的特点促使作者对其开展更深入的作用机制研究。
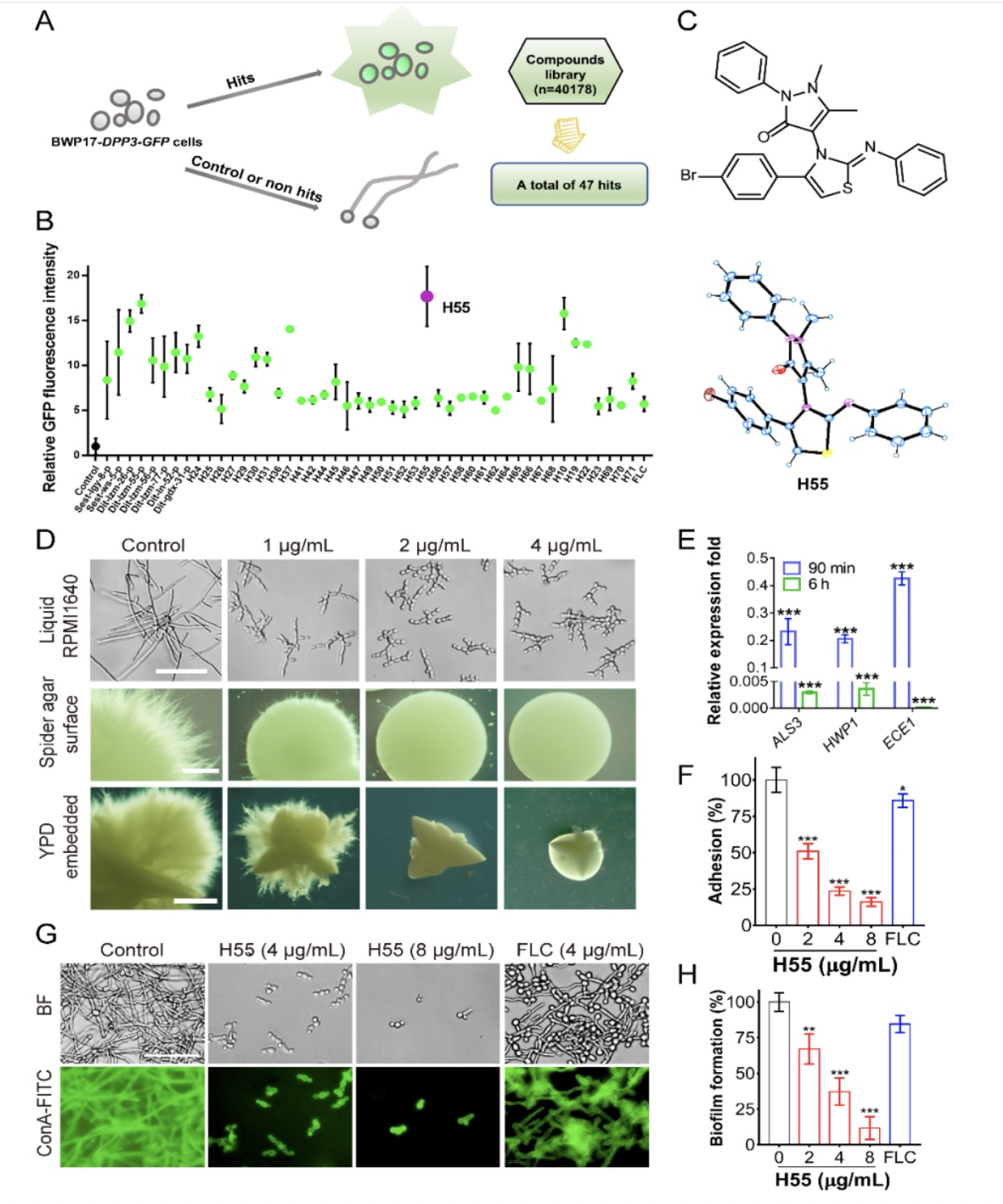
图1 Dpp3-GFP高通量筛选示意图,先导化合物H55抑制多种真菌毒力
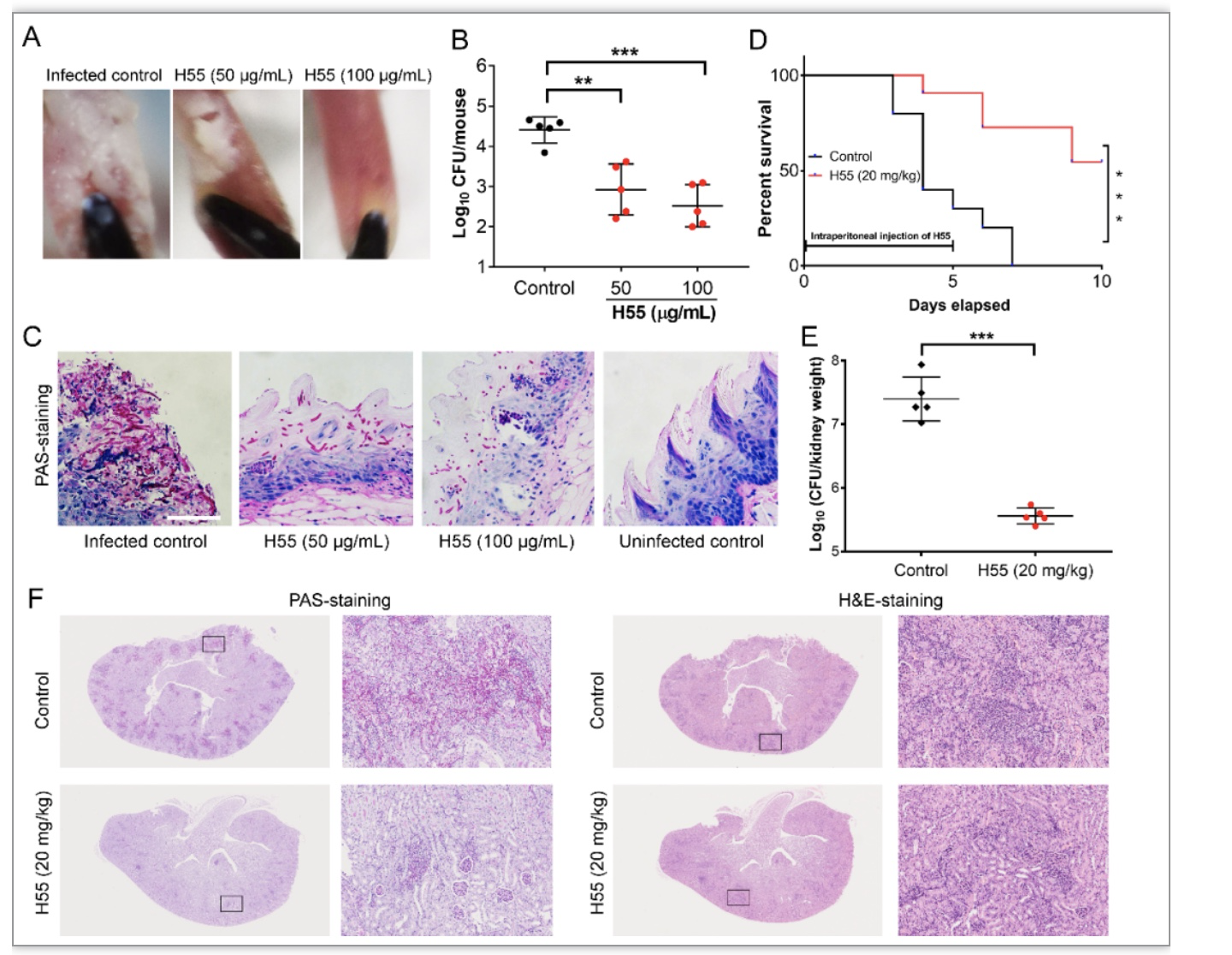
图2 H55在小鼠口腔粘膜感染模型和系统性模型中均表现出了较好的治疗效果
接下来,作者通过气相质谱联用(GC-MS)检测了H55对白色念珠菌法尼醇合成的影响,结果显示H55能够显著升高白色念珠菌法尼醇合成的水平(图3A,B,C),但H55对Dpp3敲除菌株(KWN2)的菌丝抑制能力并没有减弱(图3D),这表明H55提升法尼醇合成水平不是其发挥毒力抑制作用的主要机制。作者考虑到法尼醇合成与甾醇合成的联系,进一步通过GC-MS检测了H55对白色念珠菌甾醇合成通路的影响,由图3E可以发现,H55显著诱导了甾醇中间体zymosterol的积累,而zymosterol是C-24甾醇甲基转移酶(Erg6)的底物。许多研究已经表明Erg6的敲除会导致菌丝形成缺陷[5,6]。作者由此假设:H55通过抑制Erg6的功能发挥毒力抑制作用。作者通过提取白色念珠菌微粒体蛋白和异源表达Erg6,通过酶活测试证实了H55是Erg6的抑制剂,并通过细胞热转变分析实验(CESTA)佐证了H55与Erg6的结合(图4C,D)。
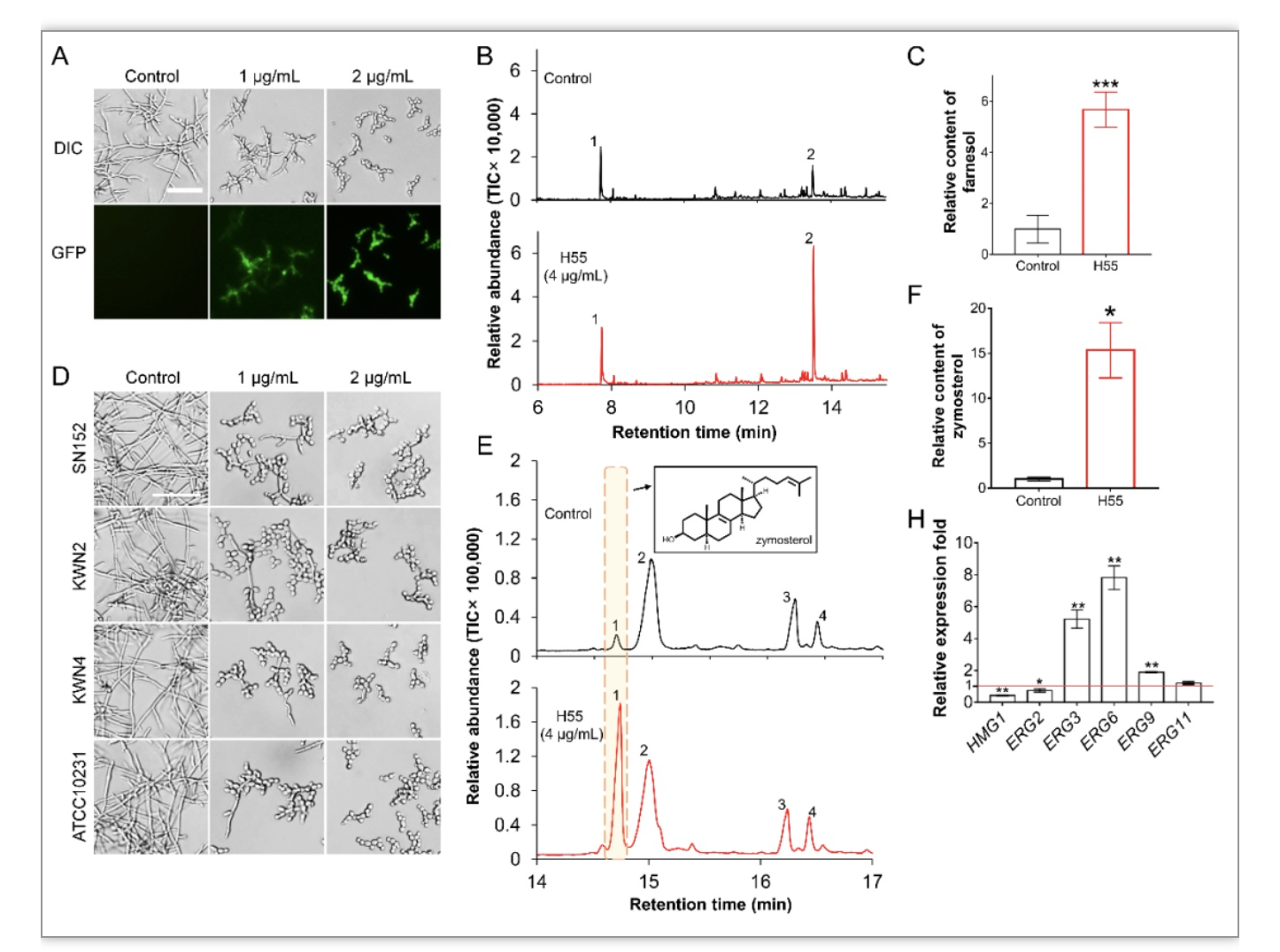
图3 H55对法尼醇合成和甾醇合成的影响
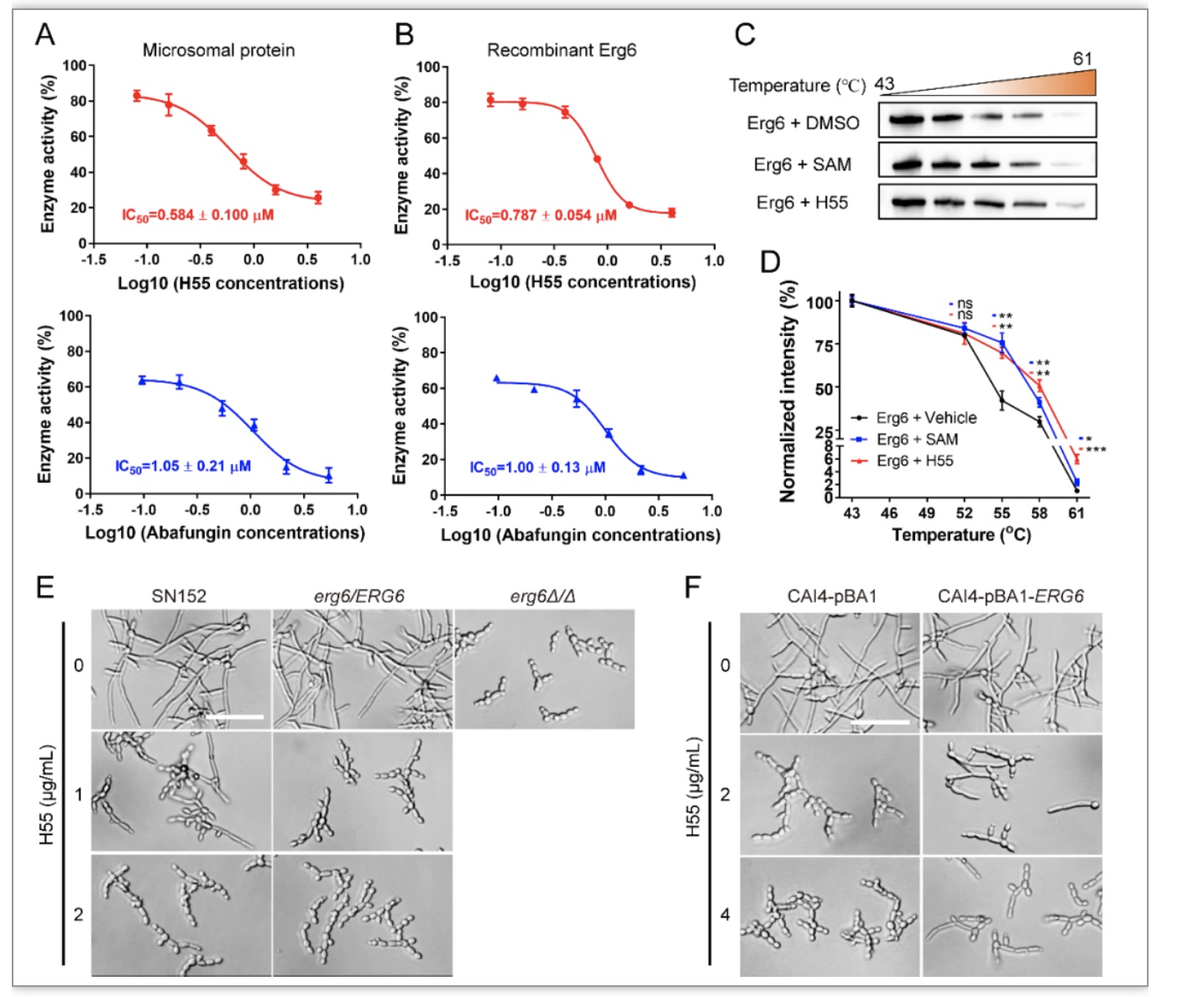
图4 H55抑制Erg6活性
为了进一步确定H55与Erg6的结合口袋,作者合成了H55的光交联探针,通过液质鉴定了一个关键区域(residues:339-356)可能与H55相互作用(图4A)。由于Erg6蛋白晶体尚未被报道且不易获得,作者首先从AlphaFold数据库中获取了预测的Erg6蛋白三维结构,然后以光交联实验结果为线索使用AlphaSpace预测了关键区域周围的可发生结合的口袋(图5B)。作者进一步使用AutoDockVina对Erg6-H55复合体进行建模。得分最高的结果表明H55和图中所指示的口袋之间存在良好的相互作用,而且预测的H55结合口袋与底物结合口袋位置不同。当该预测口袋中的关键残基K338突变为丙氨酸时,H55对Erg6-K338A的抑制作用减弱(图5C)。突变蛋白的CESTA实验结果(图5D,E,F)进一步支持所推测的结合模式。分子动力学模拟实验也证实H55在变构位点的结合降低了Erg6蛋白与底物的结合能力。综合实验结果,作者推测H55是Erg6潜在的变构抑制剂。

图5 H55是Erg6潜在的变构抑制剂
作者进一步研究了Erg6与菌丝调控通路的相关联系。实验结果显示,Erg6被抑制或敲除都会导致Ras1-cAMP-PKA通路的下调,以及菌丝特异性转录因子的表达水平改变(图6L)。
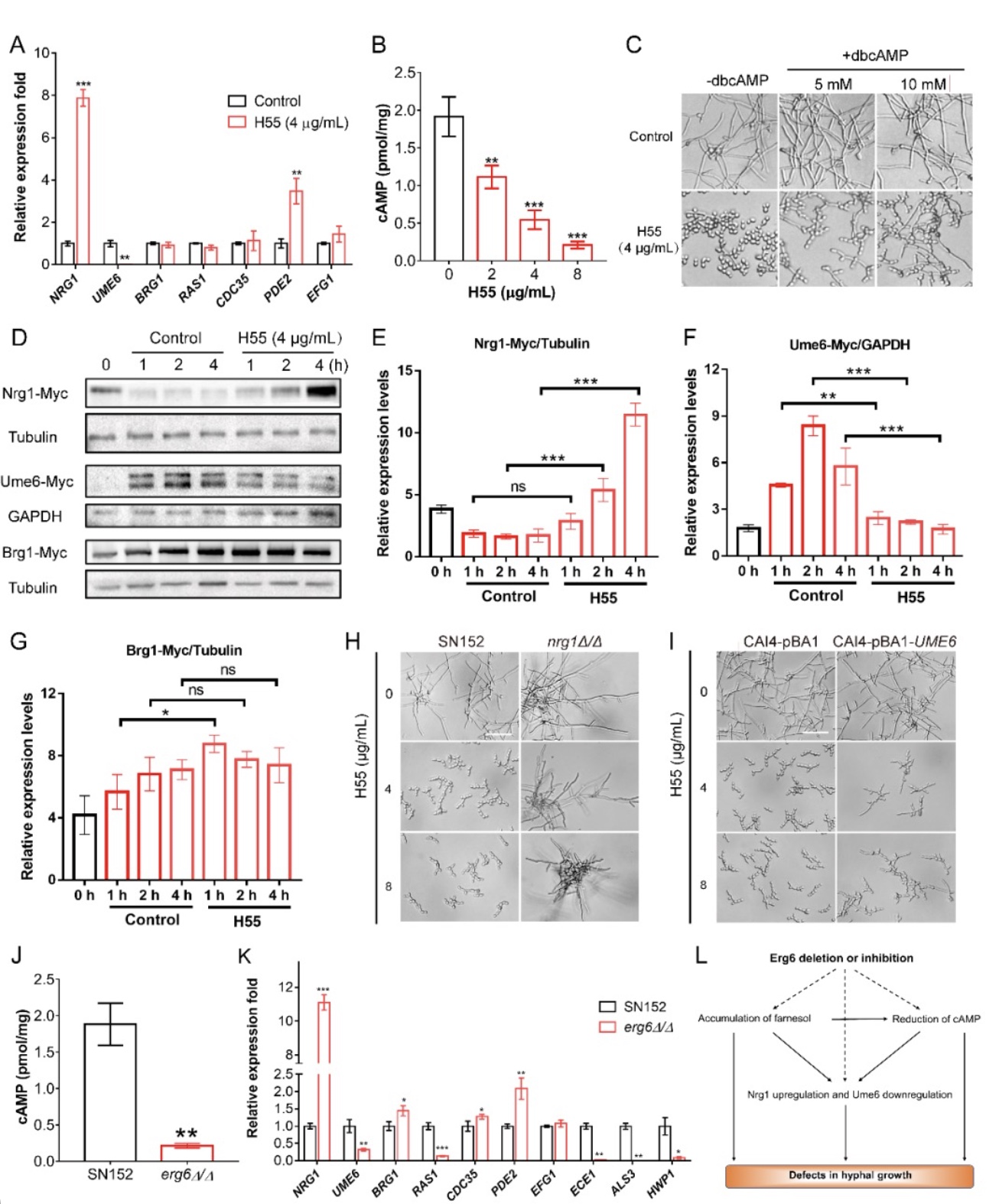
图6 Erg6对菌丝形成通路的影响
综上所述,由于真菌中麦角甾醇合成途径与哺乳动物细胞中胆固醇合成途径之间的差异,以麦角甾醇合成通路为目标是开发具有低细胞毒性的抗真菌药的重要策略。作者筛选得到了了一种安替比林类衍生物H55,它通过抑制真菌特异性酶C-24甾醇甲基转移酶(Erg6)增加了法尼醇的合成。H55作为Erg6的抑制剂,在小鼠感染模型中使白色念珠菌处于低毒力的酵母态。该工作为今后开发Erg6抑制剂来治疗真菌感染提供了新的思路。但值得注意的是,H55与Erg6的确切结合模式仍需晶体学分析来进一步佐证。
