全基因组关联研究(GWAS)已揭示了数千种与各类疾病和复杂性状相关的遗传变异。然而,多数这些与疾病相关的变异位于非编码区,其功能尚未明了。虽然非编码区的变异无法直接通过改变蛋白质编码序列来影响基因功能,但它们可以通过表观遗传机制影响基因表达,或者通过影响基因表达调控来诱导表观遗传变化。其中,DNA甲基化是一种重要的表观遗传变化,它可以通过改变转录因子的结合能力、引发染色质结构变化或调控microRNA表达来影响基因表达。因此,通过甲基化数量性状(meQTL)定位寻找与DNA甲基化变异相关的遗传变异,对理解疾病相关的表观遗传机制和研究表观基因组间的个体差异的遗传结构至关重要。大多数现有的meQTL定位研究主要关注欧洲血统的人群,在其他人群中代表性不足,尤其非常缺乏对非洲血统人群的大规模研究。该研究通过在来自动脉病遗传流行病群组(Genetic Epidemiology Network of Arteriopathy,GENOA)的961名非洲裔美国人中进行大规模甲基化数量性状位点定位研究,填补了这一关键的知识空白。
该研究识别出320965个meCpG位点和4565,687个顺式(cis) meQTLs。该研究发现45%的meCpGs携带多个独立的meQTLs,这暗示了潜在的多基因遗传结构(polygenic genetic architecture)可能是甲基化变异的基础。该研究还发现多数cis-meQTLs与同一人群中的cis-eQTL具有共位性(colocalization)。这些识别出的cis-meQTLs解释了甲基化变异的部分比例(中位数=24.6%)。此外,与cis-meQTL相关的CpG位点介导了部分基因表达下的SNP遗传效应(中位数=24.9%)。该研究结果揭示了甲基化和基因表达的共同调控,促进了对非洲裔美国人常见疾病背后的表观遗传和基因调控的功能解读的重要一步。
大部分现有的meQTL研究集中在欧洲血统的人群中,而在其他人群特别是非洲血统的人群中的大型研究较少。由于等位基因频率、连锁不平衡模式以及不同人群中甲基化变异的遗传结构的差异,一个人群中发现的meQTLs在另一个人群中并不一定能被复现。为了填补这个关键的知识空白,该研究在GENOA项目中的961名非洲裔美国人上进行了全面的cis-meQTL定位分析,识别出了320965个meCpG位点和4565687个cis-meQTLs。该研究中识别的meQTLs成功复现了在不同祖源人群中识别的大部分meQTLs。该研究使用复制率(π1) 来有效地衡量先前研究中检测到的信号在本研究中被成功复现的比例。与之前的meQTL研究相比,该研究发现GENOA在非洲血统的样本中有更高的复现率(0.98)。相比之下,与欧洲血统样本相比,GENOA的复现率在0.77到0.93之间;与南亚血统样本相比,GENOA的复现率在0.91到0. 93之间。这可能是因为非洲血统的样本与GENOA样本有更相似的祖先背景和连锁不平衡结构。
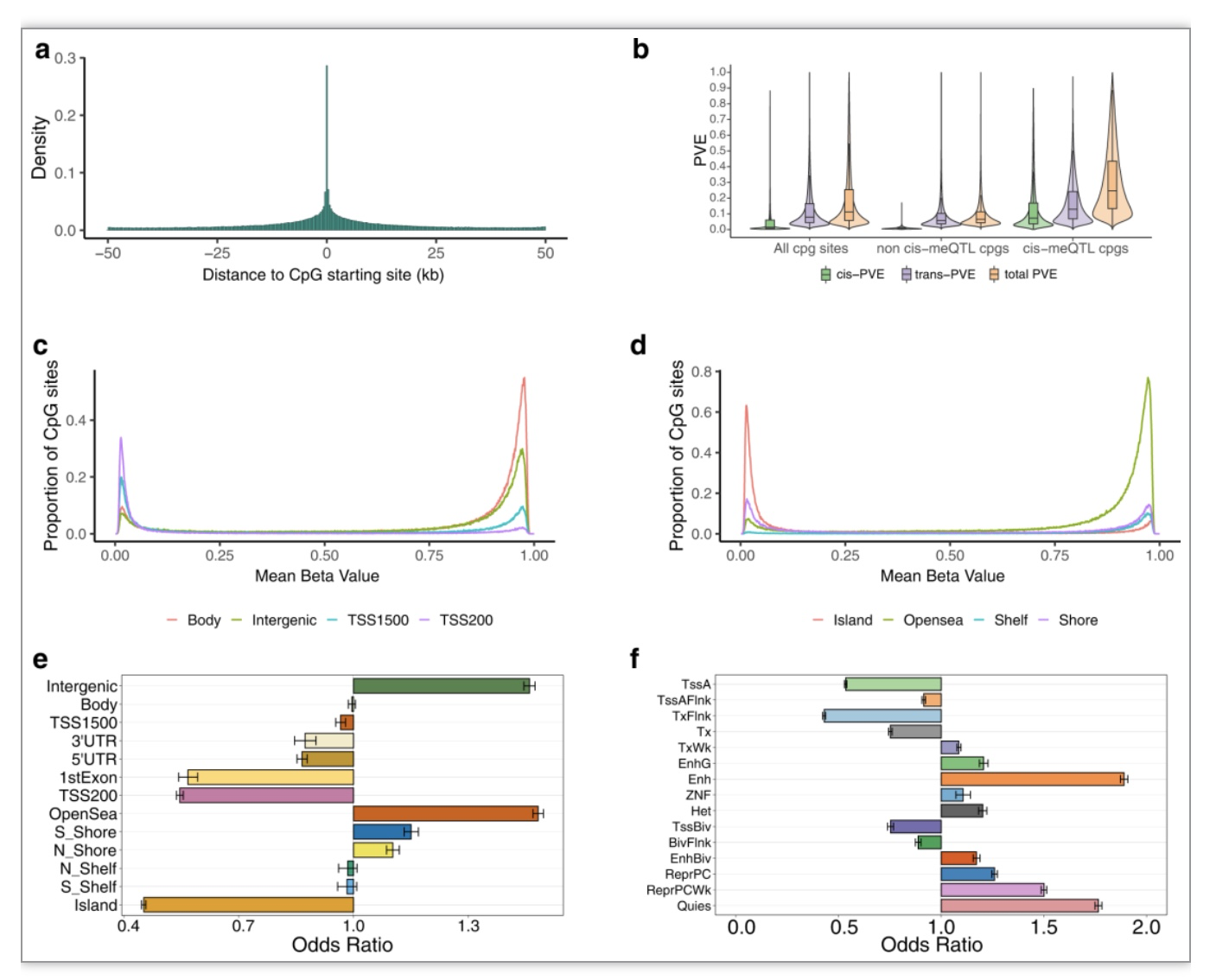
该研究进行了功能分析,以探查meCpG位点的功能特性以及它们在特定功能基因组区域的富集情况。结果发现,meCpG位点在基因间区域显著富集;然而在TSS1500、TSS200、5’UTR、第一个外显子以及3’UTR区域中则显著稀缺(如图1e所示)。此外,meCpG位点在Open Sea、North 和 South Shores、不同增强子区域以及弱转录区域中富集;而在CpG岛、各种启动子区域以及强转录区域中则相对稀缺(如图1f所示)。
(图源:Shang et al., Nature communication, 2023)
该研究通过遗传率估计和分割来研究甲基化水平变异的遗传结构。该研究使用贝叶斯稀疏线性混合模型(BSLMM)为每个CpG位点估计了所有SNPs可以解释的甲基化水平的方差比例(PVE, 即SNP heritability)。结果发现,所有CpG位点的PVE中位数为11.24%。如预期,meCpGs的PVE往往比非meCpGs的更高,其中meCpGs的PVE中位数为24.64%,而非meCpGs的PVE中位数只有6.57%(见图1b)。大部分甲基化水平的PVE是由反式(trans)SNPs解释的,只有一小部分由顺式(cis)SNPs解释。由于主要的meQTLs只能解释meCpGs中68.62%的cis-PVE,该研究进行了条件分析(conditional analysis),并使用前后步进回归(forward-backward stepwise regression)寻找独立于主要meQTL的其他meQTL。这一过程共找到了609,992个独立的meQTL,其中包括289,027个在已识别的主要meQTL基础上的条件meQTL。该研究发现大多数meCpGs(54.97%)只包含一个独立的meQTL。相当大的一部分meCpGs(23.21%)包含两个独立的meQTL,剩余的meCpGs包含三个或更多的独立meQTL(21.82%)。条件meQTL与主要meQTL相比离CpG位点更远,但与非meQTL相比,它们仍在CpG位点附近富集。通过使用主要meQTL和条件meQTL,解释的cis-PVE比例(87.35%)比仅使用主要meQTL解释的比例更高。
该研究探索了是否存在共享的遗传变异影响基因表达水平和甲基化水平。通过共定位分析,研究评估了同一个SNP对基因表达和甲基化同时产生相关影响的可能性。研究发现,被测试的基因和甲基化位点配对中有相当大的一部分(46.3%)共享一个影响基因表达和甲基化的共同SNP。在所有被测试的基因和甲基化位点配对中,有53%的配对中SNP对甲基化和基因表达的影响是相反的,并且这种相反的影响在共定位的配对中稍微多一些(55.4%)。在共定位的配对中,那些对基因表达和甲基化产生相反影响的SNP的CpG位点富集在启动子区域。此外,该研究对共定位的基因和甲基化配对进行了中介分析,以进一步检查共享的遗传变异通过一个性状影响另一个性状的程度。为此,该研究进行了两种类型的中介分析,即DNA甲基化介导了遗传变异对基因表达的影响(SNP-Methylation-Expression,SME)和基因表达介导了遗传变异对DNA甲基化的影响(SNP-Expression-Methylation,SEM),并确定了至少有111个基因和甲基化配对在至少一个方向上有显著的中介证据(FDR<0.05)。
本研究在961名非洲裔美国人中进行了全面的meQTL定位研究,揭示了甲基化变异背后的全面遗传架构。共定位和中介分析的结果也为非洲裔美国人的甲基化和基因表达共同调控提供了支持性证据。总的来说,该研究的结果代表了揭示DNA甲基化和基因表达之间共同调控的重要步骤,有助于整合和解释影响非洲裔美国人疾病病因的表观遗传和基因调控变化的功能。在未来的研究中,可以使用高维度中介分析框架构建模型来分析多个CpG位点或多个SNP,以考虑CpG位点间甲基化的相关性和由于连锁不平衡导致的SNP之间的相关性。此外,本研究的甲基化数据来自外周血白细胞,这些白细胞包括一组密切相关的细胞类型,因此,未来的甲基化数据解卷积可以识别出细胞类型特异的meQTL。通过进行trans-meQTL定位的扩展分析,将有助于全面描绘甲基化背后的遗传架构。最后,收集更多不同血统人群的甲基化数据将帮助我们更好地理解不同人群中甲基化变异背后的多样性遗传架构。