
动脉粥样硬化(Atherosclerosis, AS)是一种慢性炎症血管疾病,是导致多种心脑血管疾病的高危因素。内皮功能紊乱是AS早期的病理基础。临床研究显示,AS病灶好发于血管弯曲、分叉和狭窄部位,这些部位管腔内血液环境为以振荡切应力(Oscillatory shear stress, OSS)为特点的扰动流(Disturbed flow, DF);病灶少发于血管平直段,管腔内血液环境是以层流切应力(Laminar shear stress, LSS)为主的脉动流(Pulsating flow, PF)的血管平直段[1]。内皮细胞衬于血管最内侧,直接感应血流产生的剪切应力作用。PF有助于血管内皮细胞维持稳态并行使正常功能,而DF则会诱使内皮功能紊乱,诱发炎症反应和过度增殖,进而促使AS形成[2]。陆续有研究发现新的力学敏感因子参与内皮响应血流切应力的过程,这其中涉及的具体分子调控机制有待进一步探究。因此,揭示DF致使内皮功能紊乱促AS的力学生物学分子机制,将有助于为AS的早期诊断和治疗策略提供理论支撑。
该研究探讨了Ten-eleven-ten 1 short form(TET1s)蛋白在血流切应力诱使内皮功能紊乱促AS形成过程中的作用以及相关机制,首次揭示了TET1s为响应血流切应力调控的力学敏感蛋白。

本研究采用了体内小鼠颈动脉分支结扎模型探究不同血流切应力下TET1s的表达情况。小鼠颈动脉分支结扎后,在左颈总动脉(LCA)内形成了局部振荡切应力(OSS)环境,而在对侧右颈总动脉(RCA)则维持层流切应力(LSS)环境。检测结果显示小鼠颈动脉TET1s的mRNA和蛋白表达量在OSS环境中呈现明显的下调(图1A-B)。为排除体内其他因素影响,作者采用体外平行平板流动腔系统模拟单一流体环境,进一步确认了OSS抑制血管内皮细胞TET1s的表达(图1C)。为探究TET1s与AS形成的关系,本研究构建了基于ApoE-/-背景的TET1s敲除小鼠,通过高脂饲喂建立AS模型。由于TET1s蛋白氨基酸序列C端与全长TET1完全重合[3],因此本研究采用了TET1单敲(Tet1cs/cs)以及TET1、TET1s双敲小鼠(Tet1-/-)进行对比。高脂饲喂8周后,取小鼠主动脉进行检测观察。主动脉根部油红染色结果显示,同时敲除TET1和TET1s的实验组(Tet1cs/csApoE-/-)病灶处的脂质沉积情况远高于仅敲除TET1的对照组(Tet1-/-ApoE-/-)和空白组(ApoE-/-)(图1D-E)。以上结果提示TET1s参与了OSS引起的AS形成。
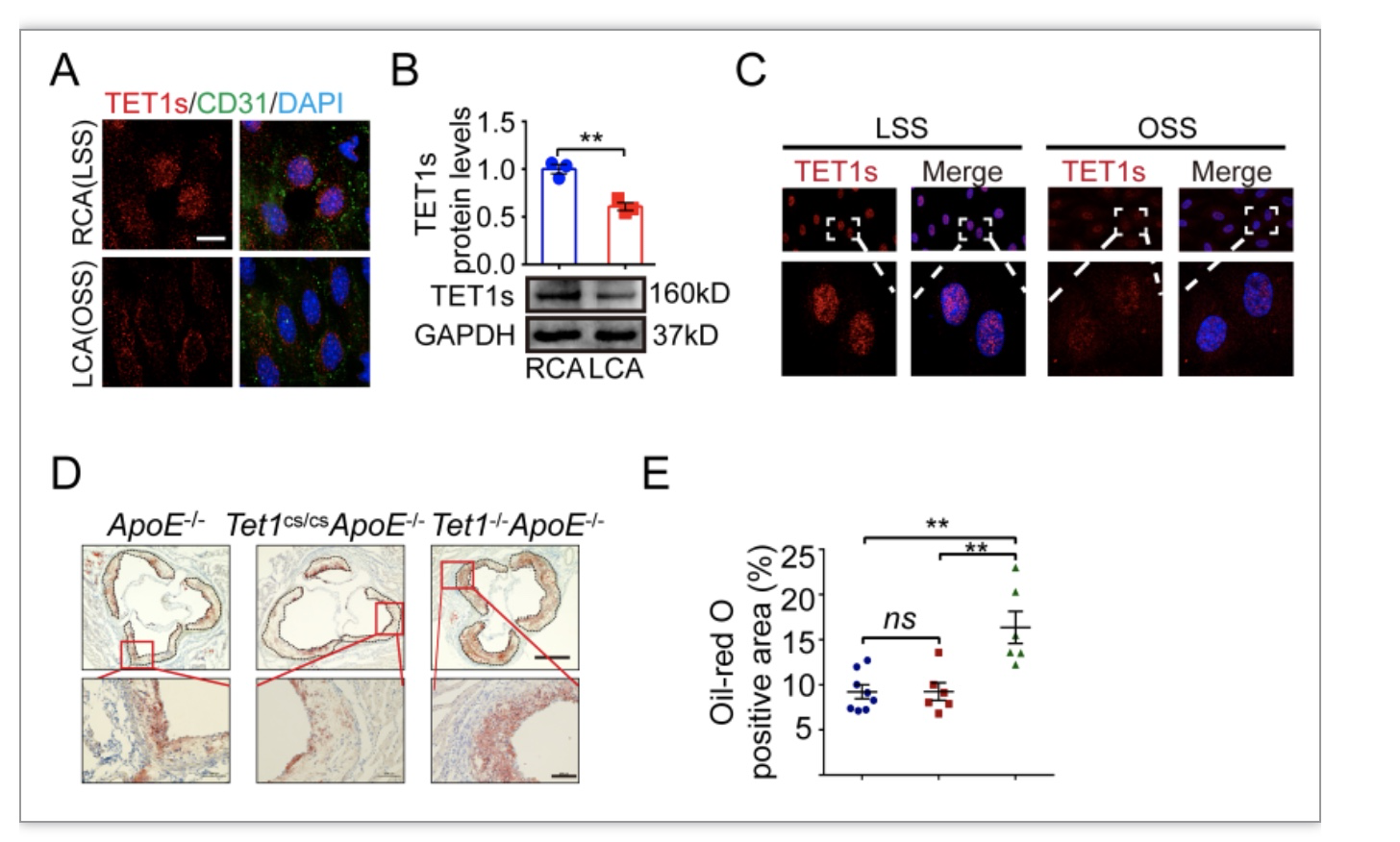
图1 不同切应力下TET1s的表达情况及TET1s与AS形成的关系
本研究接着通过功能获得和功能缺失实验,结合体外力学处理,探究TET1s在血管内皮细胞中的具体功能。使用siRNA敲低TET1s表达后发现,内皮细胞数量明显增加,增殖相关标志物的表达显著上调(图2A-B),而使用腺病毒感染细胞过表达TET1s后,细胞增殖受到抑制(图2C)。与此同时,过表达TET1s抑制了OSS诱使的细胞增殖标志物高表达(图2D)。此外,作者还发现敲低TET1s引起了血管细胞粘附分子ICAM-1、VCAM-1的高表达,进而也促进了脂多糖(lipopolysaccharide, LPS)诱导的炎症状态下单核细胞THP-1在内皮细胞上的附着(图2E-F)。当过表达TET1s时,内皮细胞中的粘附分子表达以及LPS诱导的THP-1细胞粘附都得到显著抑制(图2G)。以上结果证明在OSS环境中,TET1s通过抗增殖、抗炎症发挥对内皮细胞功能的保护作用。
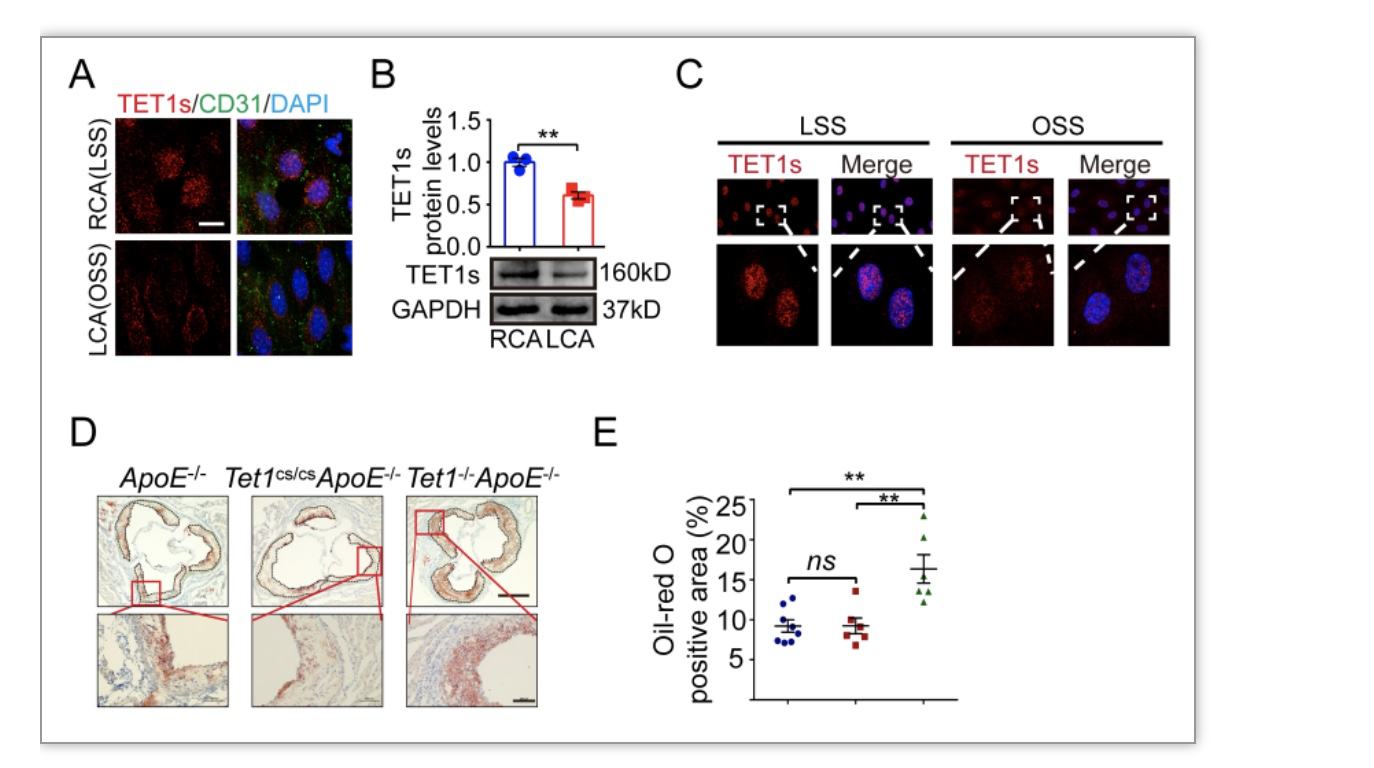
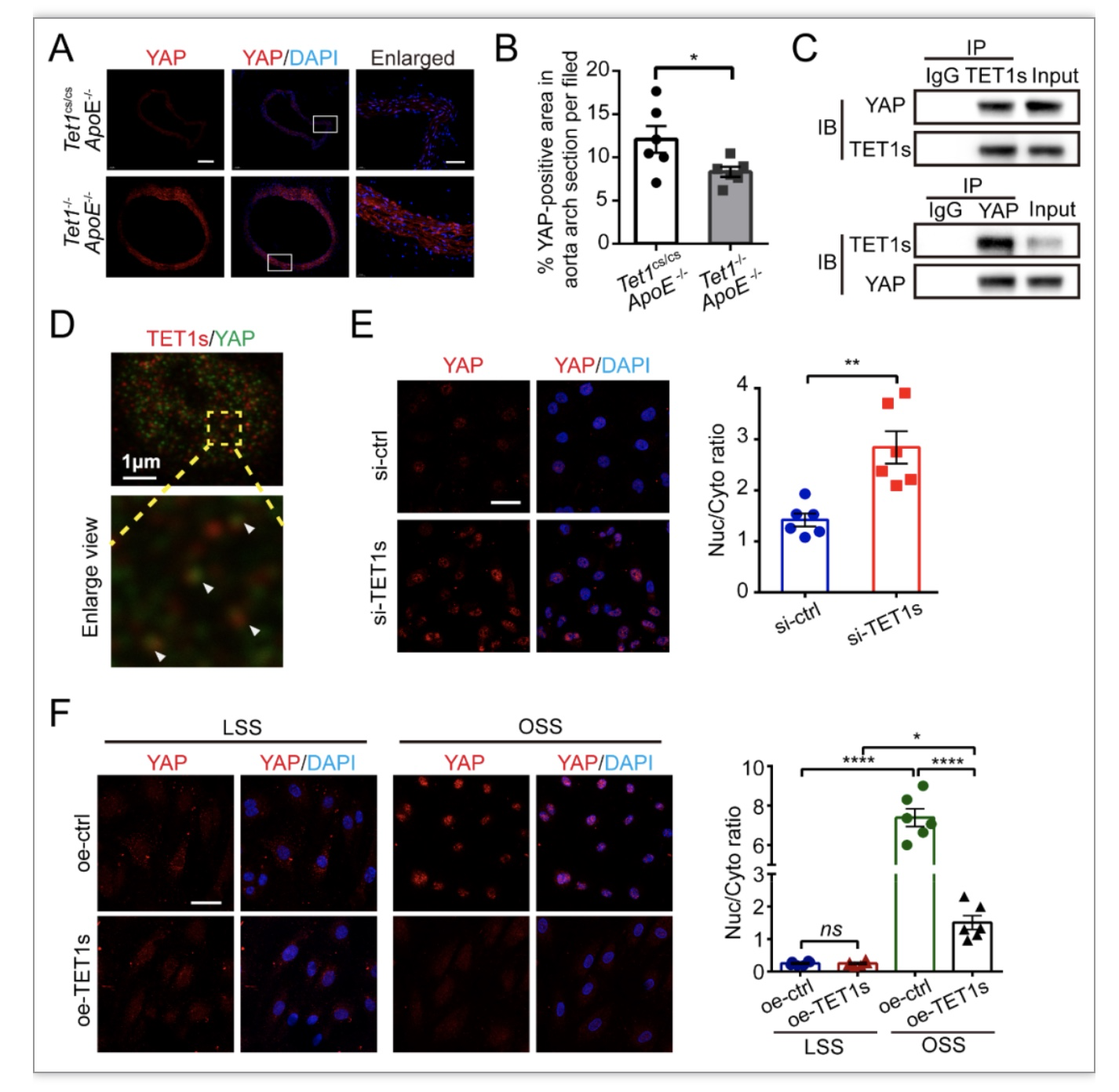
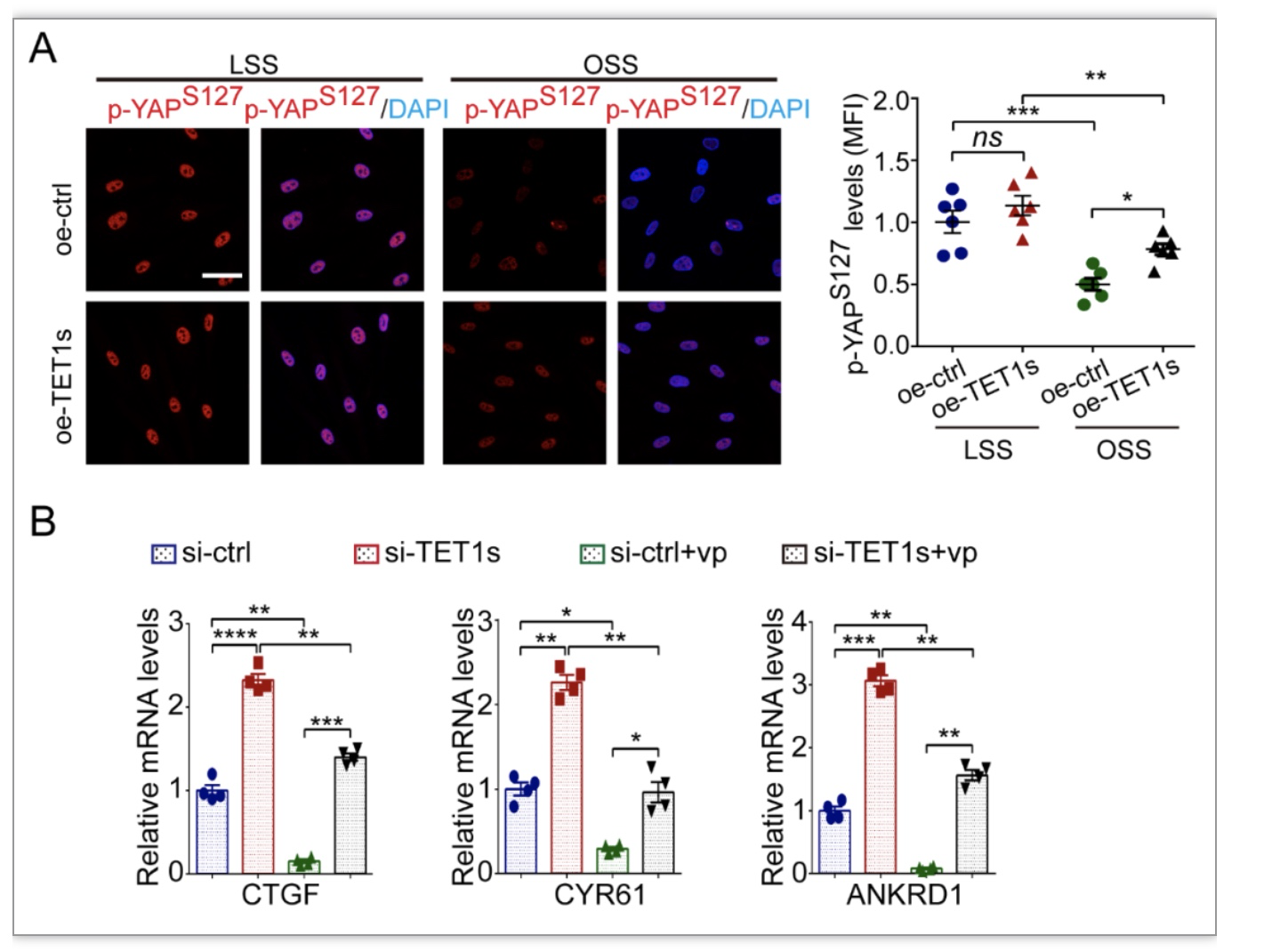
综上所述,本研究发现TET1s作为一个全新的流体切应力敏感蛋白,可响应血流刺激调控血管内皮细胞进而参与AS形成过程。具体来说,TET1s通过提升YAP蛋白第127位丝氨酸残基的磷酸化来调控YAP的活性,进而抑制内皮增殖和炎症相关基因的表达,保证血管内皮细胞在血流环境中行使正常功能。本研究的发现将为AS的临床预防和治疗策略提供一定的理论依据。现阶段的研究,初步确立了TET1s与YAP的联系,而在动态力学环境下,TET1s如何影响YAP活性、TET1s与YAP空间互作等具体分子机制还值得继续深入研究。
