
该研究提供了一种更高效且更长作用时间的抗肝细胞癌(hepatocellular carcinoma, HCC)的ASO纳米制剂,同时揭示了中性胞苷/阳离子混合脂质递送结构优化的ASO的功能和机制,具有良好的临床应用潜力。

CT102是一种靶向人胰岛素样生长因子1型受体(IGF1R)基因的磷酸化脱氧寡核苷酸,能有效抑制HCC细胞的增殖。为进一步提高CT102的抗肿瘤效果,本研究进一步使用了gamper修饰策略。CCK8结果(图1A)显示,通过增加糖环2'-位修饰位点,抗肿瘤活性增强。其中CT102MOE5对靶mRNA的基因沉默水平显著高于CT102,达到约80%的基因沉默(100 nM,图1B)。凋亡实验显示(图1C-D),CT102MOE5显著增强了CT102诱导晚期凋亡的能力,是CT102的近两倍(57% vs 31%)。因此,确定CT102MOE5为最优gapmer候选物,并进行后续的末端分子缀合。末端缀合选取了不同的糖基分子,包括乙酰氨基半乳糖(G3/Gal)、乙酰氨基葡萄糖(Glu)和甘露糖(Man)等多种衍生结构。通过进一步的体外细胞评价发现,糖基缀合结构可以一定程度上提高药物的细胞摄取,这可能是细胞表面糖基化受体导致的。此外,肿瘤表现出增强的糖摄取特性,以维持其细胞的快速增殖,这有利于增加肿瘤细胞对药物的特异性内吞作用[12]。因此,部分缀合结构在抗增殖能力及靶基因沉默方面有进一步的提升。值得一提的是,本研究发现虽然缀合结构可以在一定程度上促进细胞摄取,但其基团大小会影响ASO进入细胞核的能力。ASO在细胞核中的分布的减少可能会降低其整体活性的发挥。Ionis制药公司同样也报导了与本研究类似的G3缀合结构GalNAc3-ASO用于HCC的治疗。然而,随后的体内验证表明,缀合物与非缀合物相比没有显着差异,而本研究发现的ASO在胞内定位的变化可能是造成活性无法进一步提升的一个重要原因[13]。
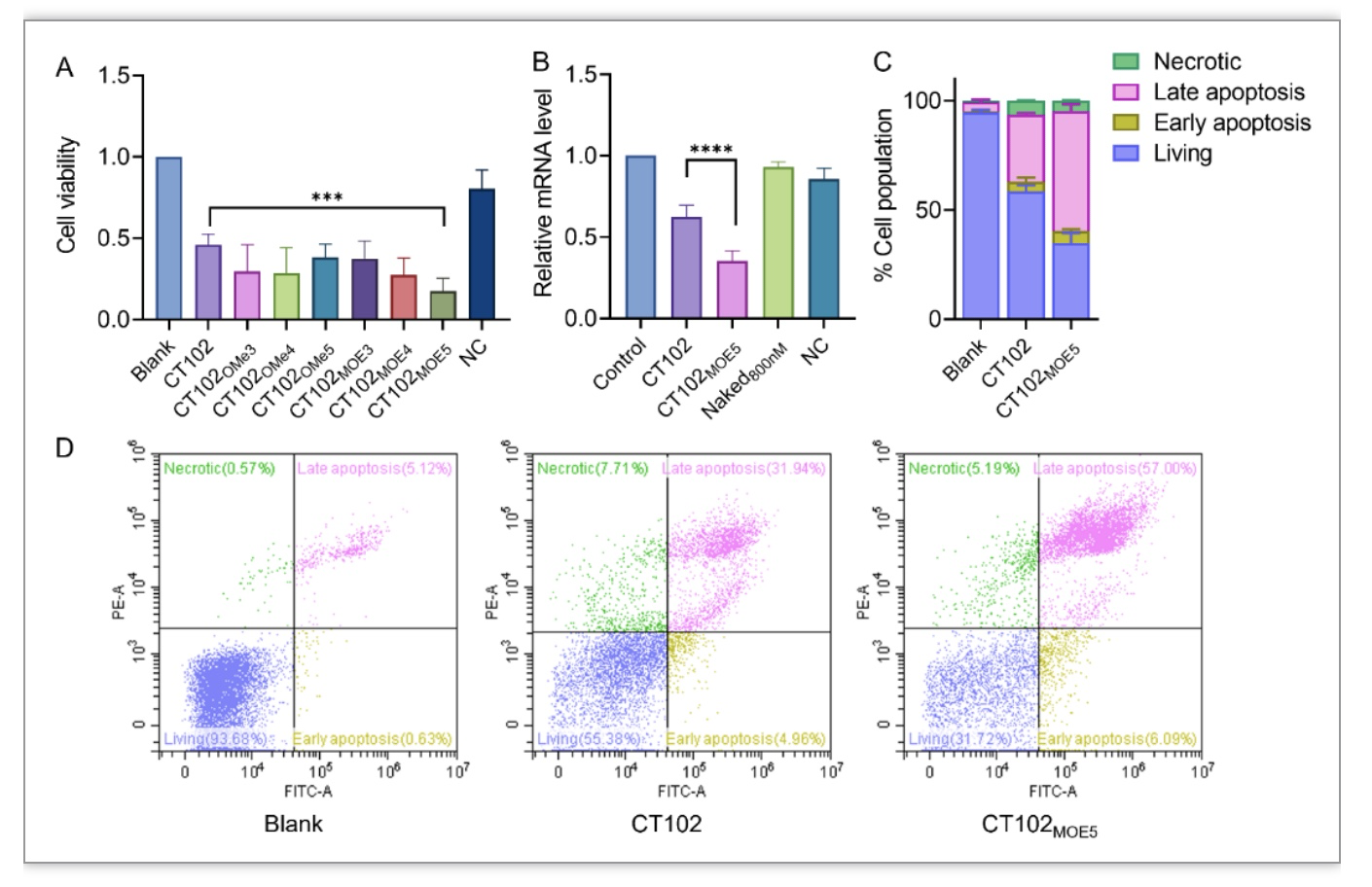
图1. CT102不同gapmer修饰物在HepG2细胞中的抗肿瘤活性
小鼠体内肿瘤生长28天结果(图2A-B)显示,与索拉非尼和CT102相比,CT102MOE5和其它几种缀合物的抗肿瘤效果有显著差异,并且缀合物组(G3/Gal/Glu/Man-CT102MOE5)优于CT102MOE5组,第28天的平均肿瘤大小分别是第0天的15.0、9.4、7.5和18.2倍(CT102MOE5-24.6倍)。在所有给药组中,Glu-CT102MOE5的抑瘤作用最强,停药后1周内肿瘤生长倍数变化最小,仅为1.1倍。随后,进行肿瘤内靶基因敲除的定量评估和IGF1R蛋白的免疫荧光分析。结果显示,靶标IGF1R mRNA的沉默(图2C)与蛋白表达的变化一致(图2D-E)。gapmer CT102MOE5进一步增强了CT102的体内活性,有效地沉默了IGF1R mRNA的表达水平(P<0.001),并降低了随后的蛋白表达(P<0.01)。与CT102MOE5相比,G3-CT102MOE5在基因和蛋白水平上没有优势。但与CT102MOE5和G3-CT102MOE5相比,Glu-CT102MOE5进一步增强了基因沉默,抑制了蛋白表达水平(P<0.05)。总体而言,活性数据结果显示,Glu-CT102MOE5显著抑制肿瘤生长,被确定为抗HCC的最佳活性结构。
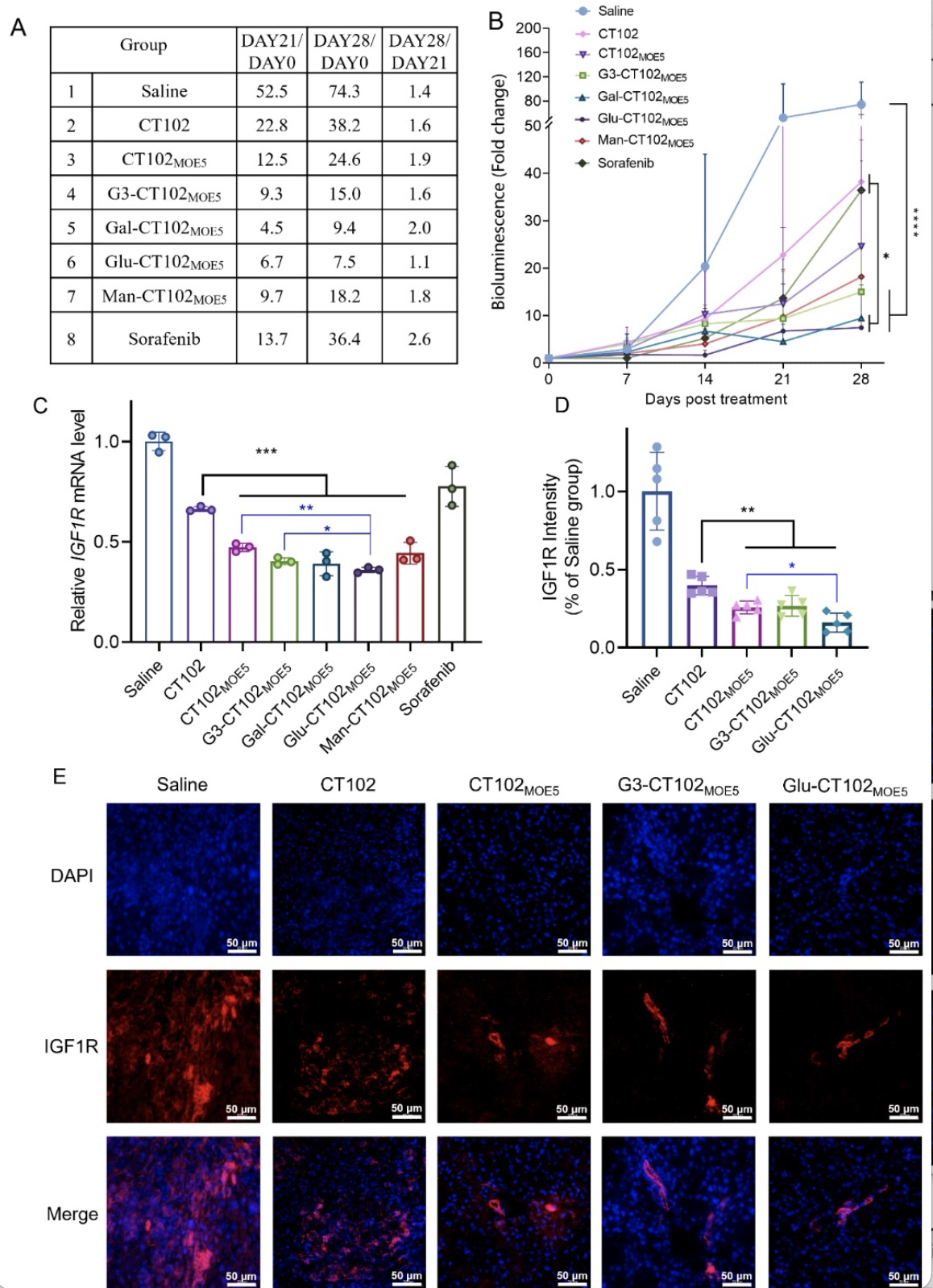
图2. 不同结构优化的CT102/Mix制剂在原位肝癌小鼠模型中的抗肿瘤药效
CT102及其衍生物引起生物学变化的作用机制被进一步研究。Western blotting检测IGF1R、AKT、p-AKT、PI3K、p-PI3K蛋白表达水平。CT102、CT102MOE5和Glu-CT102MOE5给药24h后,IGF1R蛋白水平明显降低,而gapmer CT102MOE5和缀合物Glu-CT102MOE5均能有效抑制PI3K和AKT的磷酸化水平,其中Glu-CT102MOE5的抑制能力最为显著(图3A)。PI3K-AKT信号通路在许多癌症类型中异常激活,在肿瘤细胞增殖和存活中起核心作用,并且是调节细胞增殖、分化和凋亡所必需的。CT102及其衍生物(尤其是Glu-CT102MOE5)可不同程度地有效降低靶蛋白IGF1R的合成,从而抑制下游PI3K-AKT信号通路,进而抑制肿瘤发展。此外,本研究还比较了靶向IGF1R mRNA的ASO (CT102、N-04、N-06)和siRNA (siIGF1R-1、siIGF1R-2)的细胞增殖抑制和基因沉默活性(图3D)。其中,ASO N-04的靶mRNA位点与CT102的靶mRNA位点相距52个碱基,靠近外显子1的末端。ASO N-06靶向IGF1R mRNA的外显子2,该外显子与siIGF1R-1靶向位点部分重叠。siIGF1R-2的靶向位点与CT102相同。在相同浓度(50 nM)下,ASO和siRNA在HepG2细胞中的IGF1R mRNA沉默活性具有可比性(图3B),无显著性差异(~60%)。细胞增殖抑制活性结果显示(图3C),CT102 (70.9%)对HepG2细胞增殖的抑制活性明显强于N-04 (48.5%)、N-06 (42.5%)、siIGF1R-1 (21.3%)和siIGF1R-2 (22.6%)。这些结果表明CT102具有复杂的抗肿瘤活性,提示CT102不仅抑制IGF1R的表达,还可能作用于其它凋亡相关基因。

图3. CT102及其衍生物的作用机制
因此,通过体外转录组学和蛋白质组学分析来评估CT102的可能靶标。结果显示,上千个基因和蛋白质发生改变,许多信号通路被影响(图4B)。与空脂材(Mix)相比,CT102同时有13个基因和蛋白上调,5个基因和蛋白下调(图4A),差异表达基因富集多种肿瘤相关信号通路。除了已知的酪氨酸激酶受体介导的IGF1R信号转导发生变化外,PPAR、P53、DNA复制、错配修复等信号通路也存在显著差异(图4C-E)。此外,通过合理的化学修饰,gamper CT102MOE5和缀合物Glu-CT102MOE5影响的基因和蛋白数量减少,一定程度上增强了CT102的“on-target”效应。蛋白质组学结果显示,CT102、CT102MOE5和Glu-CT102MOE5组中约25%的差异表达蛋白定位于细胞核内,包括参与转录、磷酸化、磷酸酶激活和DNA/RNA结合蛋白的功能蛋白。结果发现,修饰后的CT102可以降低细胞核和细胞质中IGF1R mRNA的表达水平(图4F),并对在肿瘤发生发展中具有功能作用的GAS2、POLA2、LGALS2和IGFBP1的表达产生显著影响。因此,针对如此高比例的CT102在细胞核中的潜在机制,尤其是具体作用值得探讨。结合转录组学和蛋白质组学分析结果,CT102及其衍生物不仅在酪氨酸激酶激活的PI3K-AKT通路中发挥作用,而且在DNA复制、抗炎、免疫微环境等许多影响肿瘤生长的过程中发挥作用。它们可以通过不完全匹配与其他mRNA (如GAS2、POLA2和LGALS2)相互作用,从而下调相关蛋白的表达,共同诱导肿瘤细胞的凋亡过程。表明ASO不仅可以靶向成熟mRNA阻碍翻译并发挥沉默活性,在细胞核中的作用有多种可能,如与pre-mRNA结合、启动裂解机制、阻断pre-mRNA加工形成成熟mRNA等。也可能与细胞核内互补的双链DNA结合形成三链体[14],抑制DNA转录,或通过核酸适配体机制与转录因子或转录复合物结合,有效影响其转录[15-17],间接导致相关基因或蛋白上调,如IGFBP1。
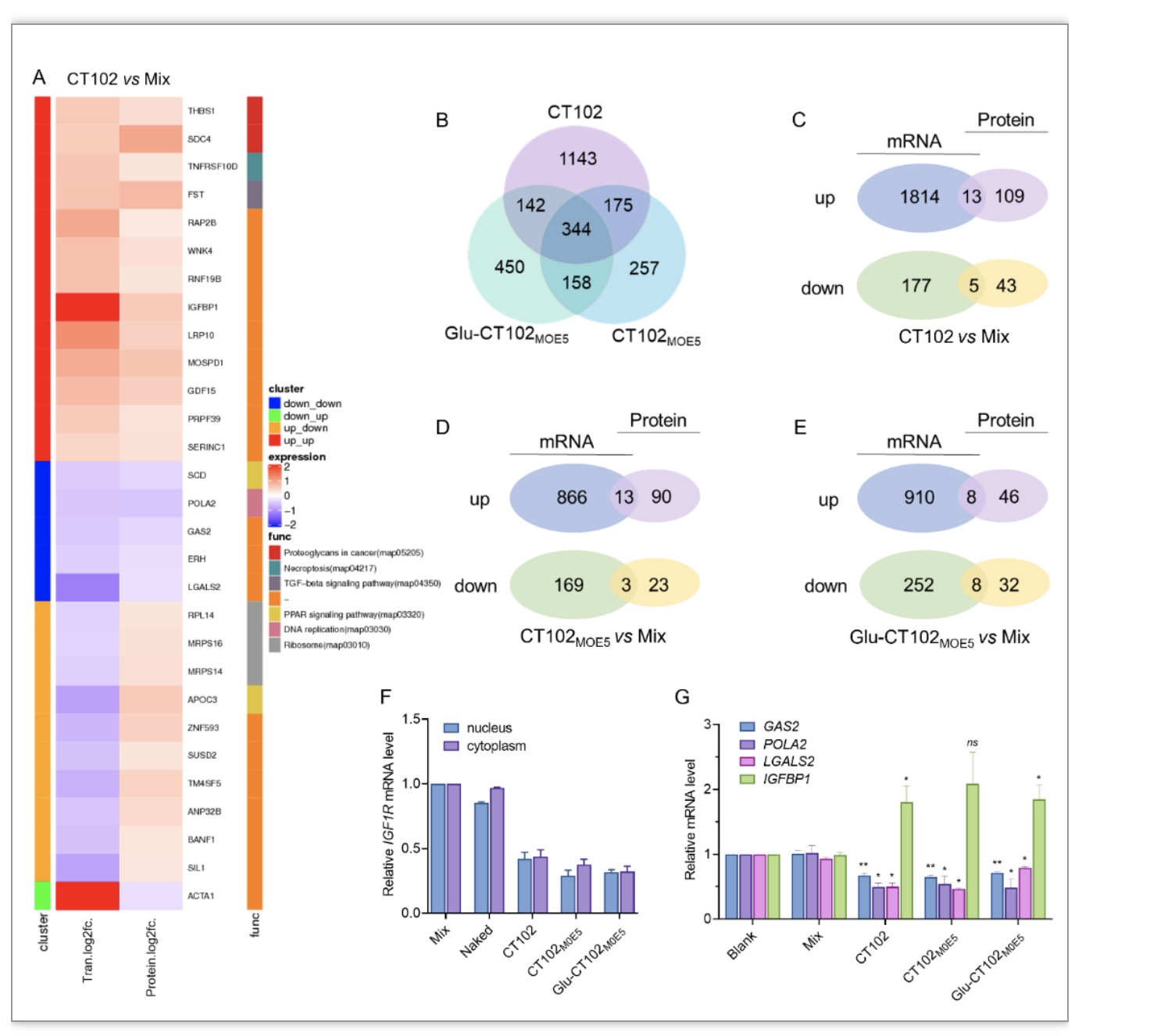
图4. CT102及其衍生物的潜在作用靶
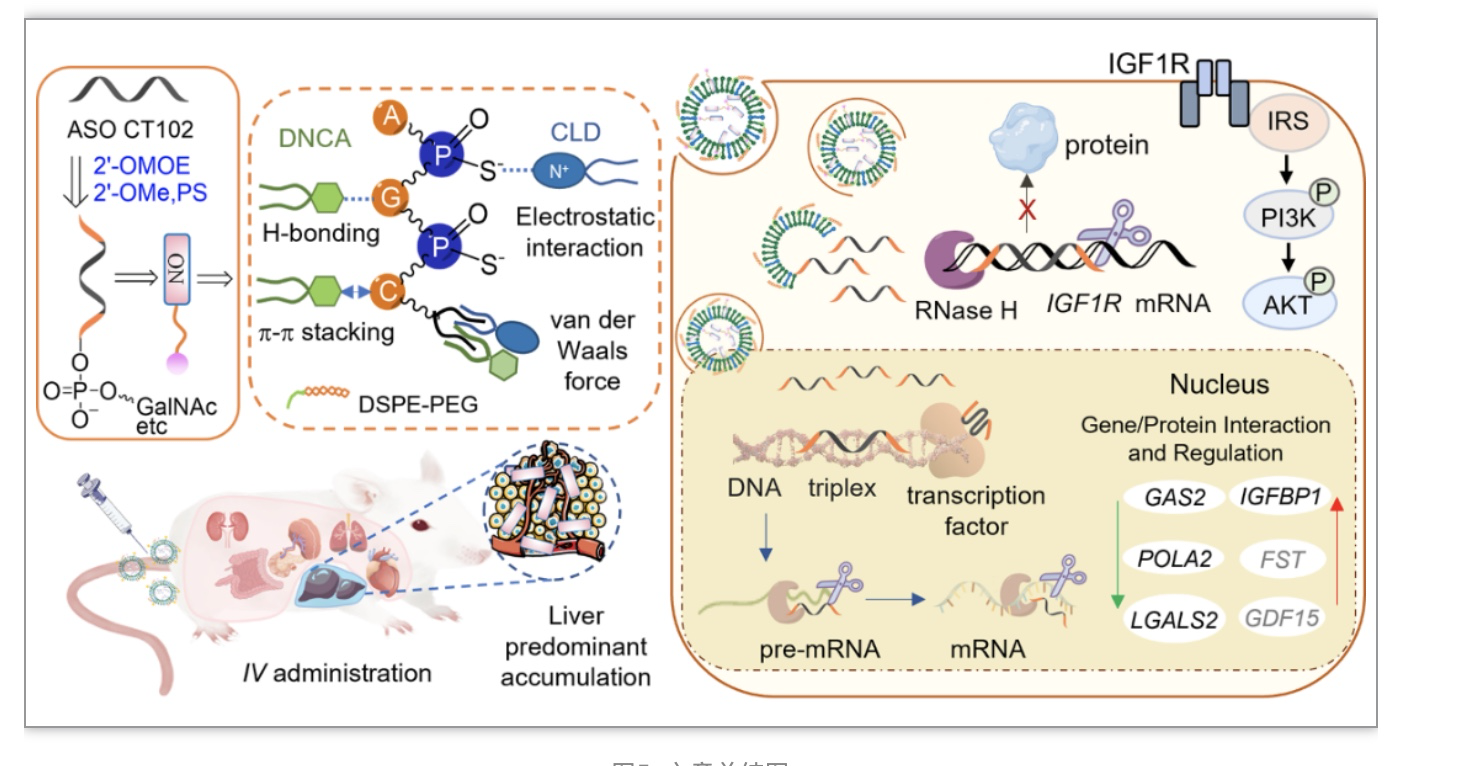
图5. 文章总结图
反义寡核苷酸(ASOs)是一类靶向mRNA或基因的治疗药物,近年来备受关注。然而,体内靶组织的有效递送和最佳蓄积仍然是一个具有挑战性的问题。CT102是一种靶向IGF1R mRNA并诱导细胞凋亡的ASO。本研究对脂质体递送下的ASO的组织分布进行了详细的探索。基于中性胞苷DNCA/阳离子CLD/DSPE-PEG混合脂质和寡核苷酸之间的多种分子间相互作用,包括氢键、π-π堆叠和静电相互作用,确定了一种高比例肝脏蓄积的药物制剂。此外,结构优化的CT102为治疗肝细胞癌(HCC)提供了一种新的策略。gapmer CT102MOE5和缀合物Glu-CT102MOE5在体外100 nM表现出较好的抗增殖和抑制IGF1R mRNA的作用,在体内低剂量、低给药频率下也有较好的效果(2mpk/4d)。转录组和蛋白质组分析表明,ASO治疗中可能同时存在其他相关靶点和功能调控。这些结果表明,脂质包封与结构优化相结合的方法在寡核苷酸药物的递送中具有良好的临床应用前景。
