
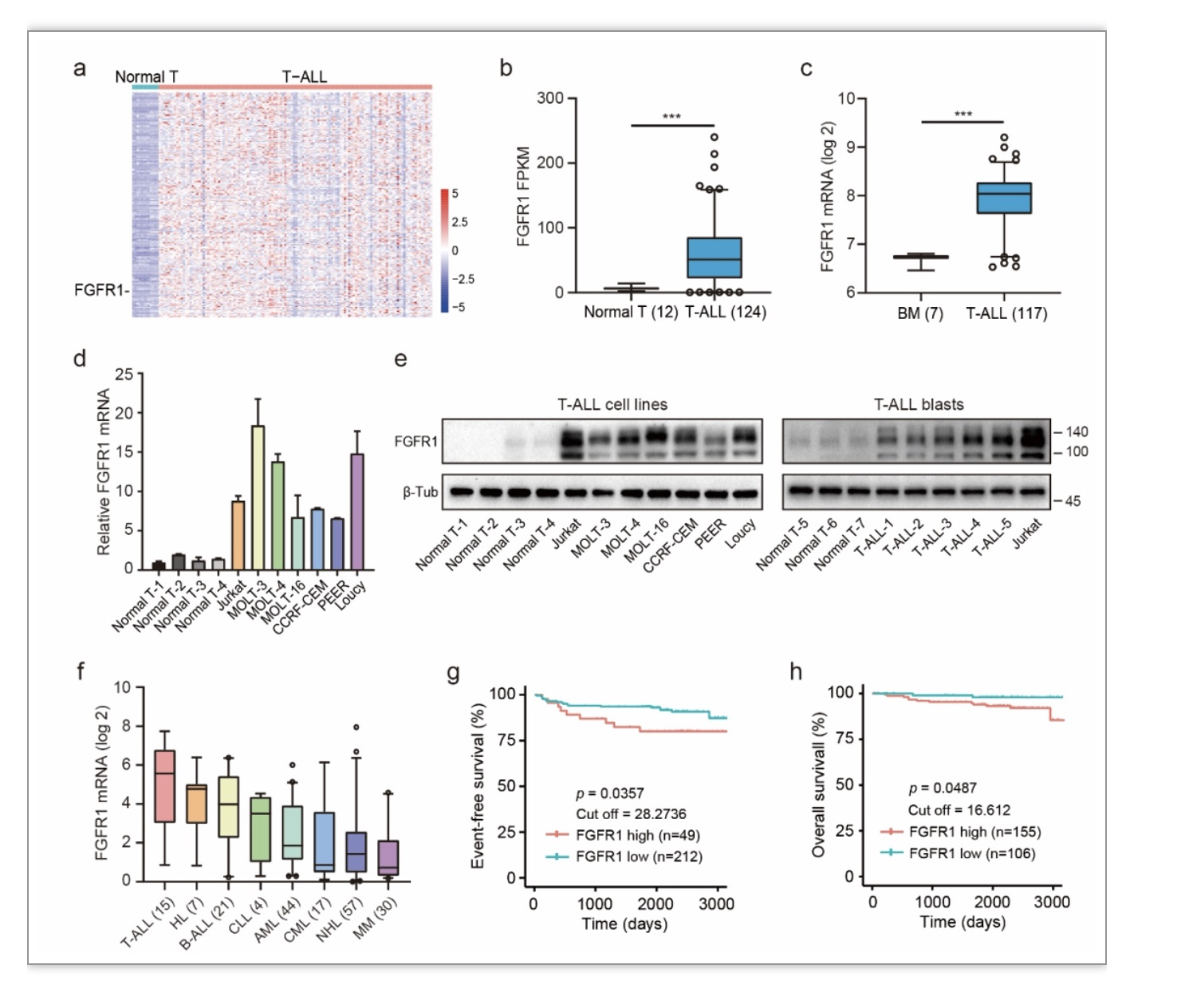
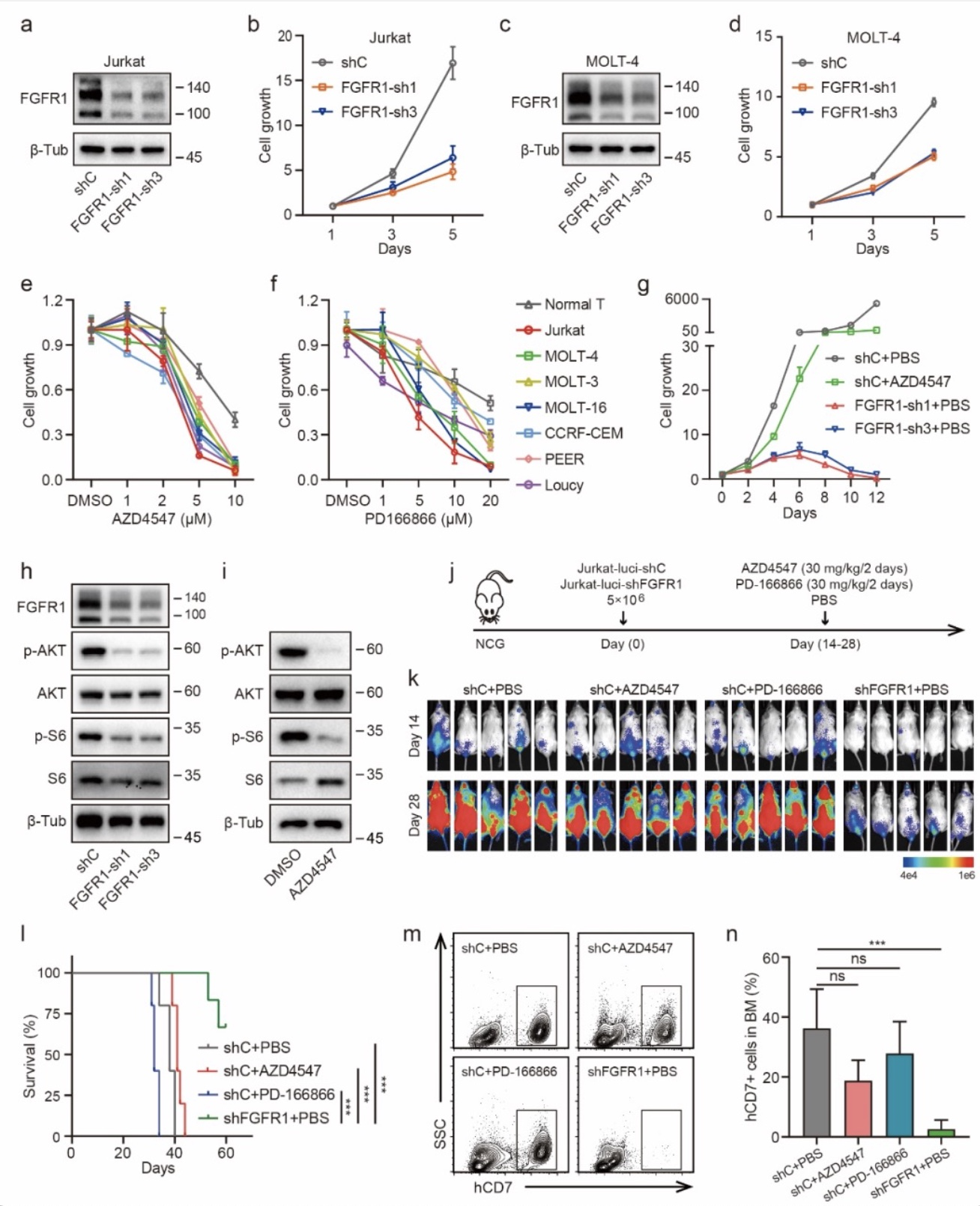
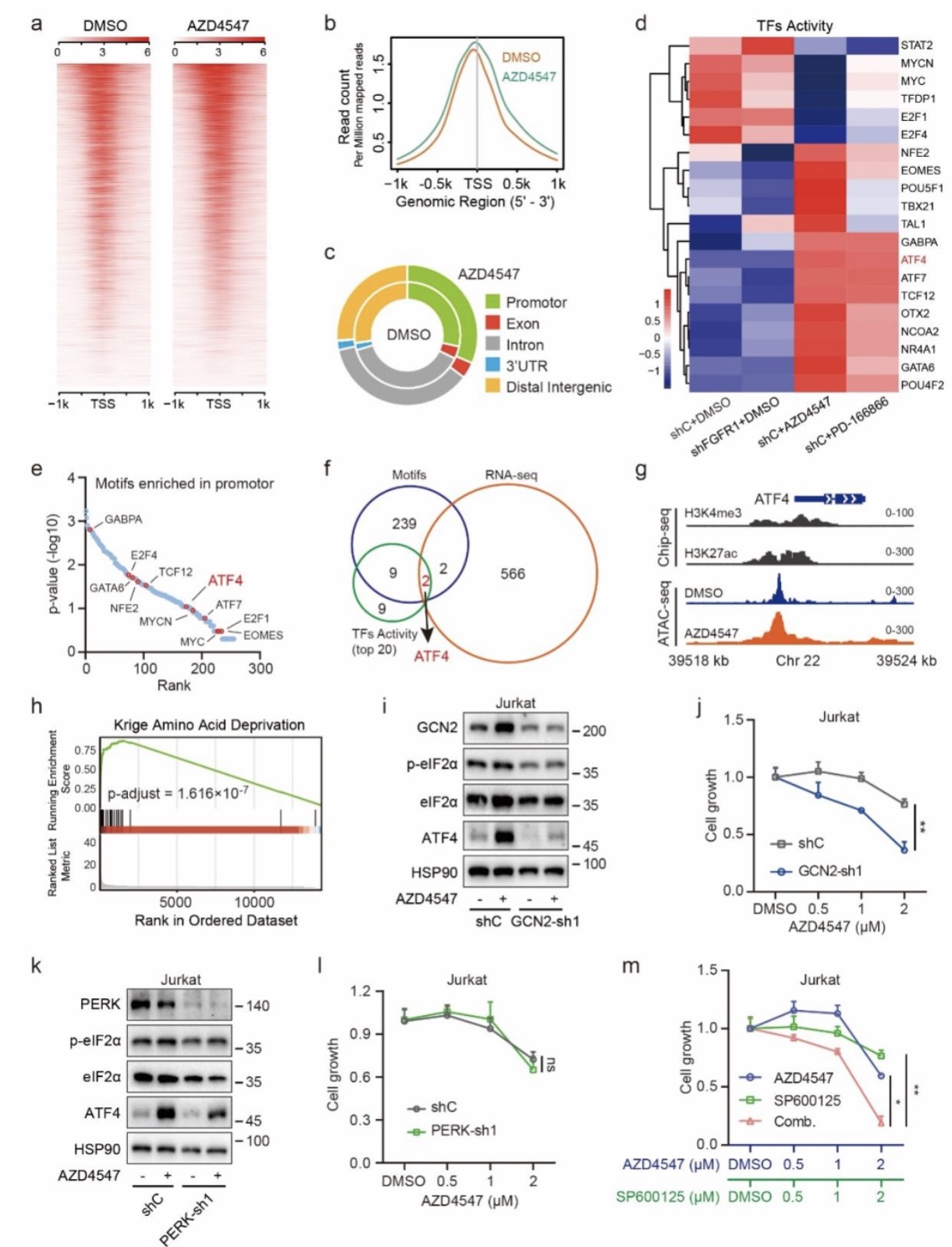
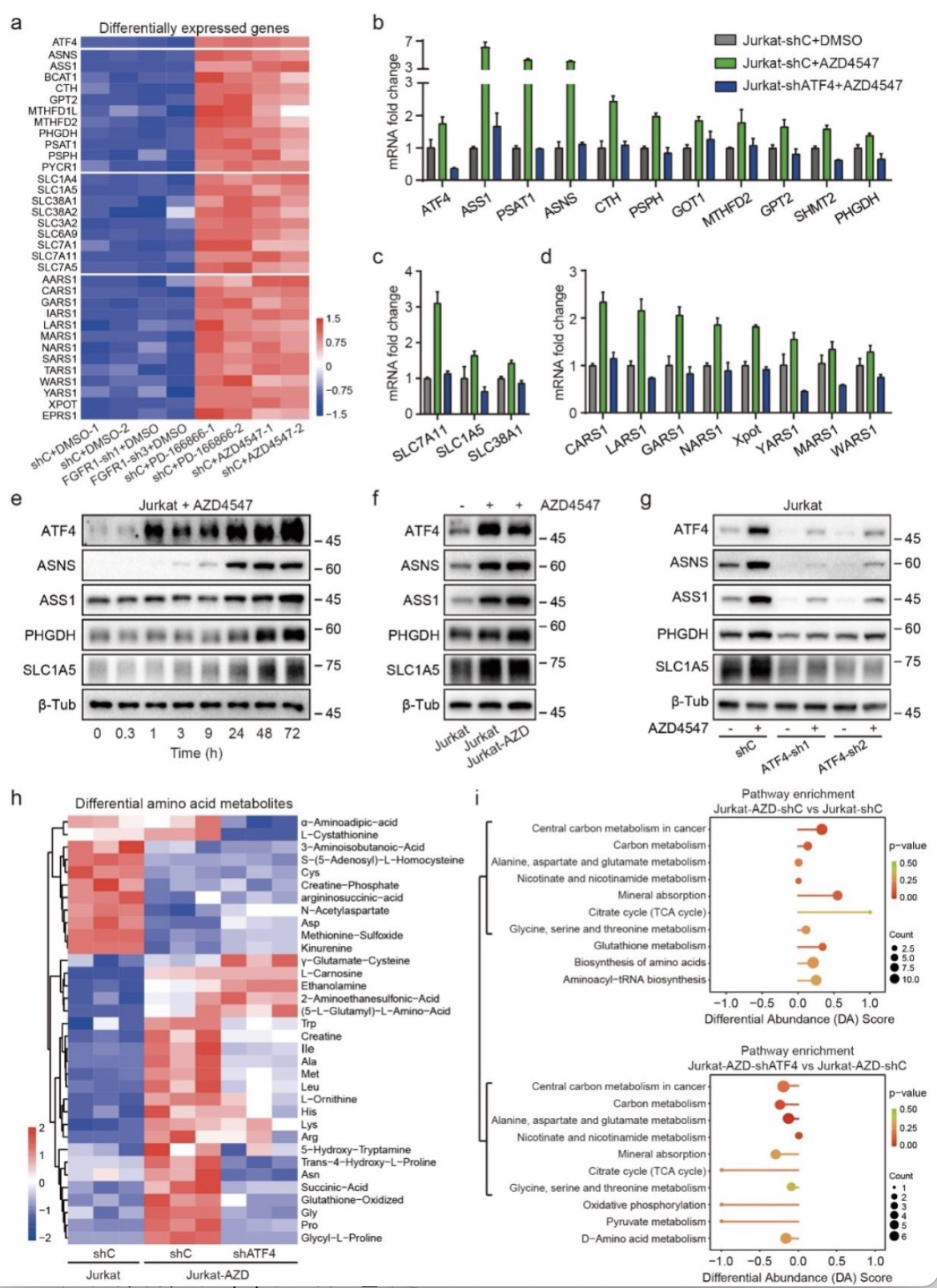
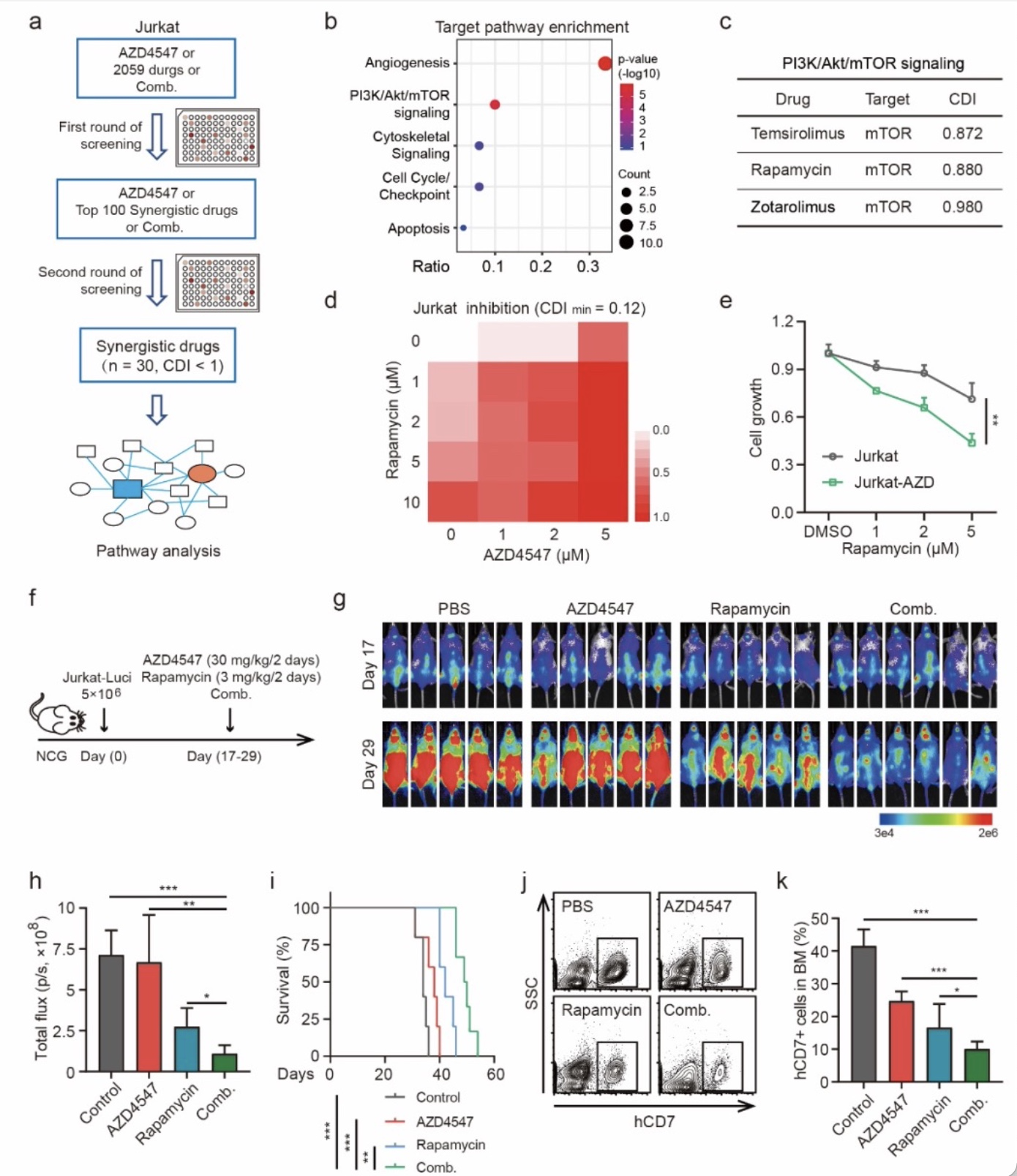
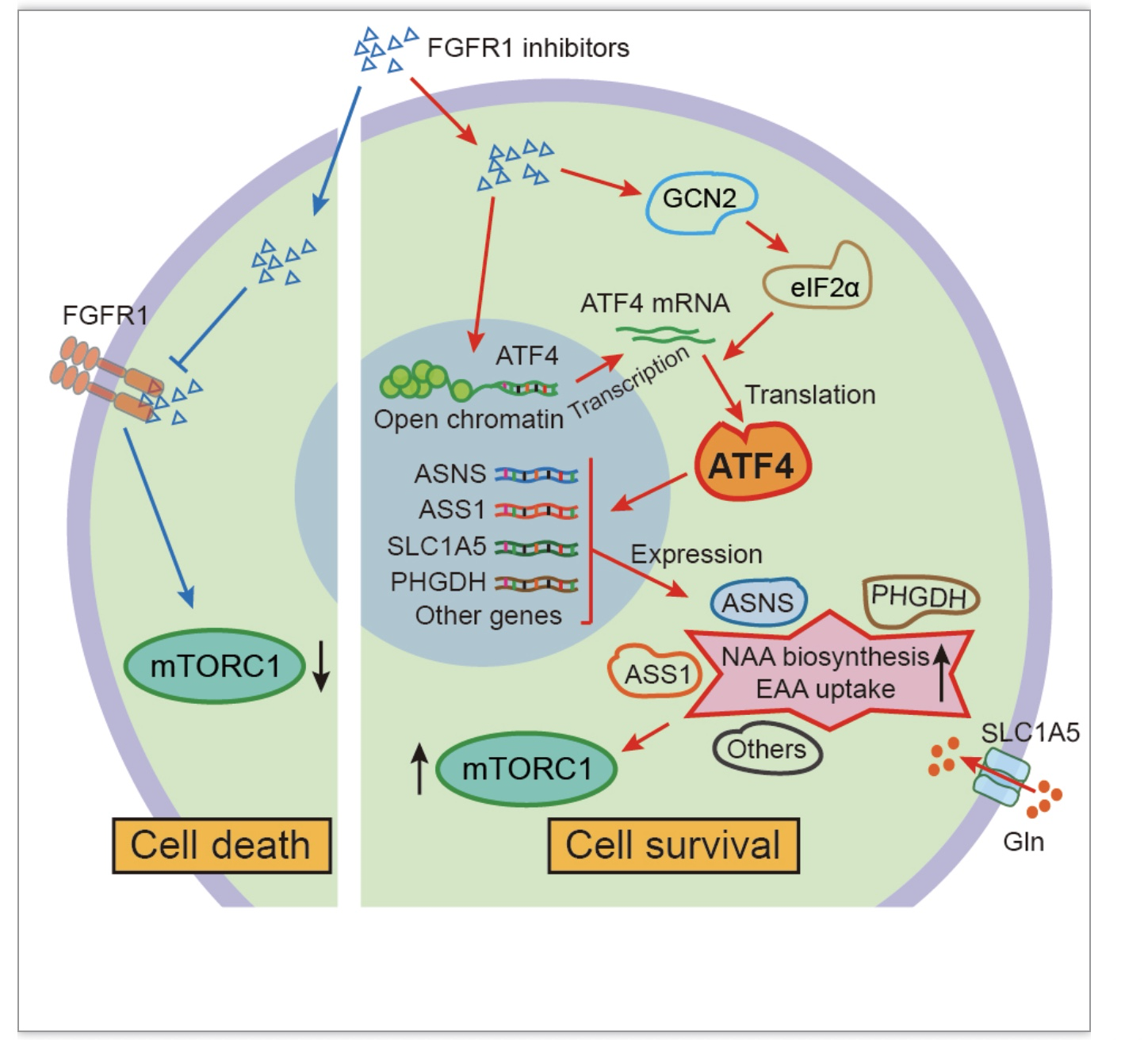
本工作首次发现FGFR1蛋白在T-ALL异常高表达且可作为T-ALL治疗的有效靶点,联合阻断mTOR通路可解决T-ALL对FGFR1抑制剂耐药的问题,并显著抑制T-ALL的进展,对FGFR的抑制剂应用于T-ALL的治疗提供了新思路。同时该研究也阐明了ATF4介导的氨基酸代谢重编程是其耐药的关键机制,后续也可尝试通过PROTAC(proteolysis-targeting chimeras)技术等技术靶向ATF4来克服耐药问题,不过还有待进一步验证。

本工作首次发现FGFR1蛋白在T-ALL异常高表达且可作为T-ALL治疗的有效靶点,联合阻断mTOR通路可解决T-ALL对FGFR1抑制剂耐药的问题,并显著抑制T-ALL的进展,对FGFR的抑制剂应用于T-ALL的治疗提供了新思路。同时该研究也阐明了ATF4介导的氨基酸代谢重编程是其耐药的关键机制,后续也可尝试通过PROTAC(proteolysis-targeting chimeras)技术等技术靶向ATF4来克服耐药问题,不过还有待进一步验证。
