
肝纤维化是肝损伤过程中的关键病理生理过程,虽然轻微的肝纤维化被认为是代偿性的,并有助于维持肝脏结构和功能,但异常的肝纤维化会加剧肝损伤并导致肝功能恶化,最终导致肝功能衰竭[1]。事实上,肝纤维化经常在慢性肝病患者中观察到,并且与预后不良有关[2]。无论肝纤维化病因如何,主流观点认为其是由肌成纤维细胞介导的[3],而肝星状细胞(HSC)是大多数体内肝纤维化模型中成熟肌成纤维细胞的主要来源[4]。趋化因子配体11 (CCL11)也称为嗜酸性粒细胞趋化因子 (Eotaxin),是CC趋化因子家族中的小分子量细胞因子[5]。大部分与 CCL11 相关的研究都集中在它作为嗜酸性粒细胞转运的调节因子的作用,相比之下,CCL11 的嗜酸性粒细胞非依赖性作用几乎没有得到充分研究。根据Berkman 等人的一项研究表明,CCL11 可以在体外直接增强人肺成纤维细胞的 ECM 生成和微观增殖能力,这一过程类似于 HSC-肌成纤维细胞转变[6],这一研究结果促使研究者提出假设:HSC源性的CCL11 可能在肝纤维化中发挥作用。
研究者首先研究了在HSCs激活过程中CCL11表达的变化。通过CCl4对C57/B6小鼠建模,分离出原代HSCs、Kupffer细胞和肝细胞,再通过定量PCR(图 1A)和ELISA(图 1B)的检测方法,都显示出CCL11在HSCs中的表达选择性和显著上调。另外,通过BDL对小鼠建模也得出了类似的结论。而从小鼠中分离出原代HSCs并在体外进行自发激活时,发现CCL11的上调与肌成纤维细胞标记基因(Acta2和Col1a1, 图1C, 1D)密切相关。接着,研究者将人肝星状细胞(LX-2)暴露于两种公认的促纤维生成生长因子TGF-β和PDGF下,两种处理均显著上调了CCL11 mRNA(图1E)和蛋白(图1F)的表达。最后,与健康对照组相比,从人类肝硬化活检标本中新鲜分离的肝细胞中观察到CCL11表达上调(图1G)。
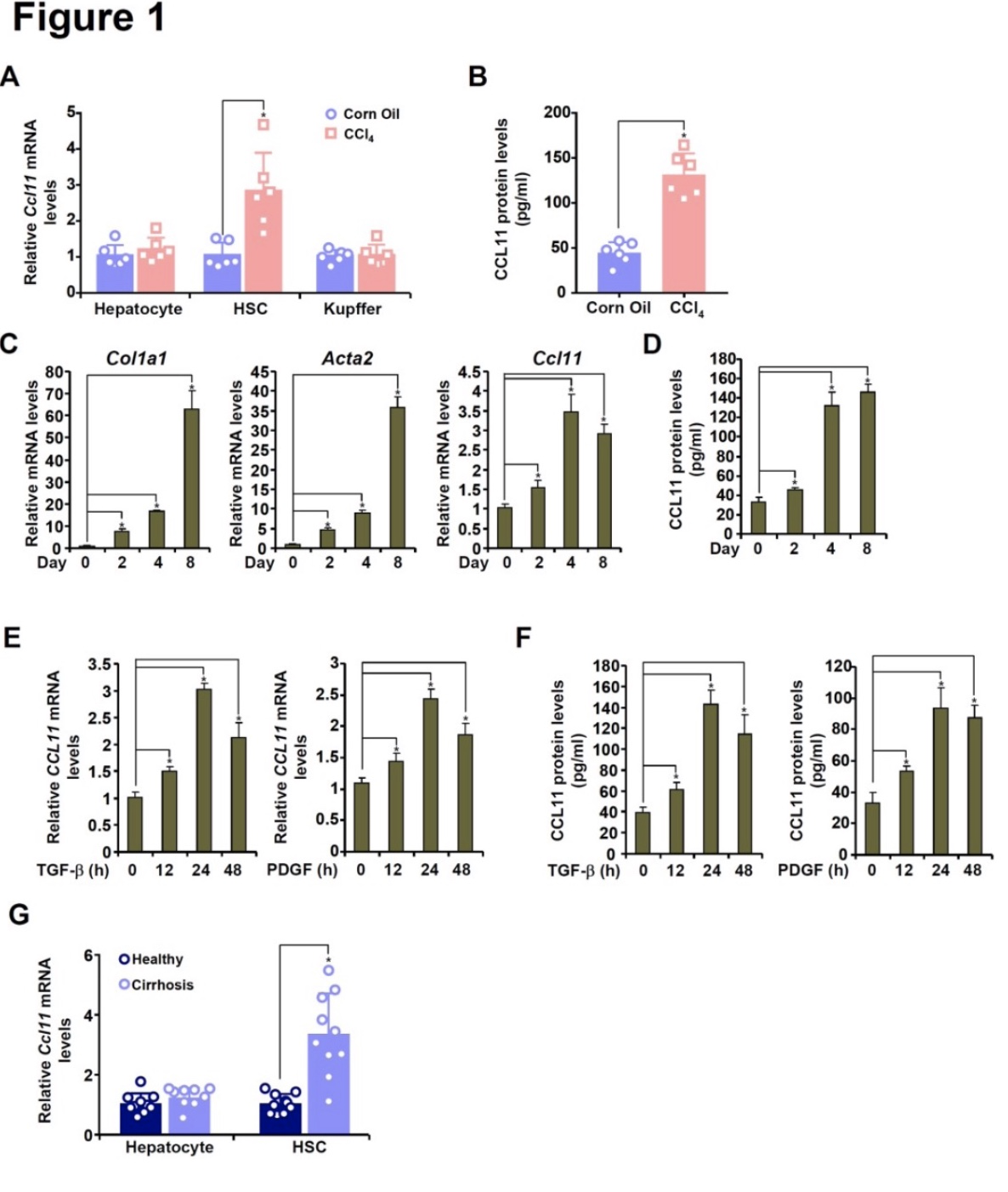
图1 CCL11的表达在HSC激活过程中上调。
(图源:M. Kong, et al, Hepatology, 2023)
研究者随后开展了关于在HSC激活过程中其诱导CCL11表达的调控机制的研究,IPA揭示了几种上游转录因子(TF)可能有助于CCL11的表达(图2A)。用siRNA单独敲掉每一个相关的TF,结果发现,在LX-2细胞中,只有ZNF281的耗尽才会导致TGF-β对CCL11诱导的明显抑制(图2B)。过表达ZNF281剂量依赖性地增强了CCL11启动子-荧光素酶报告基因的活性(图2C)。当对全长CCL11启动子-荧光素酶结构引入序列缺失时,观察到通过过表达ZNF281或用TGF-β或PDGF处理(图2E、F)激活报告基因活性需要一个相对于CCL11启动子转录起始位点-800到-400之间的结合基序。ChIP实验证实,TGF-β或PDGF处理增强了ZNF281与CCL11启动子的这一区域(-581/-449)的结合,但没有增强与更远端的CCL11启动子区域的结合(图2F)。通过TGF-β或PDGF处理,ZNF281对CCL11启动子的增强可归因于ZNF281表达本身的上调(图2G, 2H)。最后,近端ZNF281位点的突变完全消除了报告基因对TGF-或PDGF的反应(图2J)。综上所述,这些数据表明ZNF281可能介导了HSCs中CCL11的转录。

图2 ZNF281介导TGF-β诱导的HSC中CCL11的转录。
(图源:M. Kong, et al, Hepatology, 2023)
研究者接着探讨了CCL11在体外HSC激活中的相关性。首先,利用siRNA抑制内源性CCL11的表达;通过肌成纤维细胞标记基因的qPCR(图3A)和Western blotting(图3B)检测,显示CCL11缺失显著抑制了TGF-β诱导的HSCs激活。EdU实验显示,CCL11的敲除减缓了TGF-诱导的HSC增殖(图3C)。相反,重组CCL11的处理增加了HSCs中肌成纤维细胞标记基因的水平(图3D, 3E),并促进了HSCs的增殖(图3F)。当进行RNA测序来评估CCL11对HSC转录组的影响时,以p< 0.05和1.5Xfold变化作为界值,发现CCL11处理改变了HSC转录组400多个基因。GESA显示,CCL11刺激与细胞外基质重塑相关基因的上调呈显著正相关,提示CCL11可能使细胞向肌成纤维细胞样表型转变(图3G, 3H)。
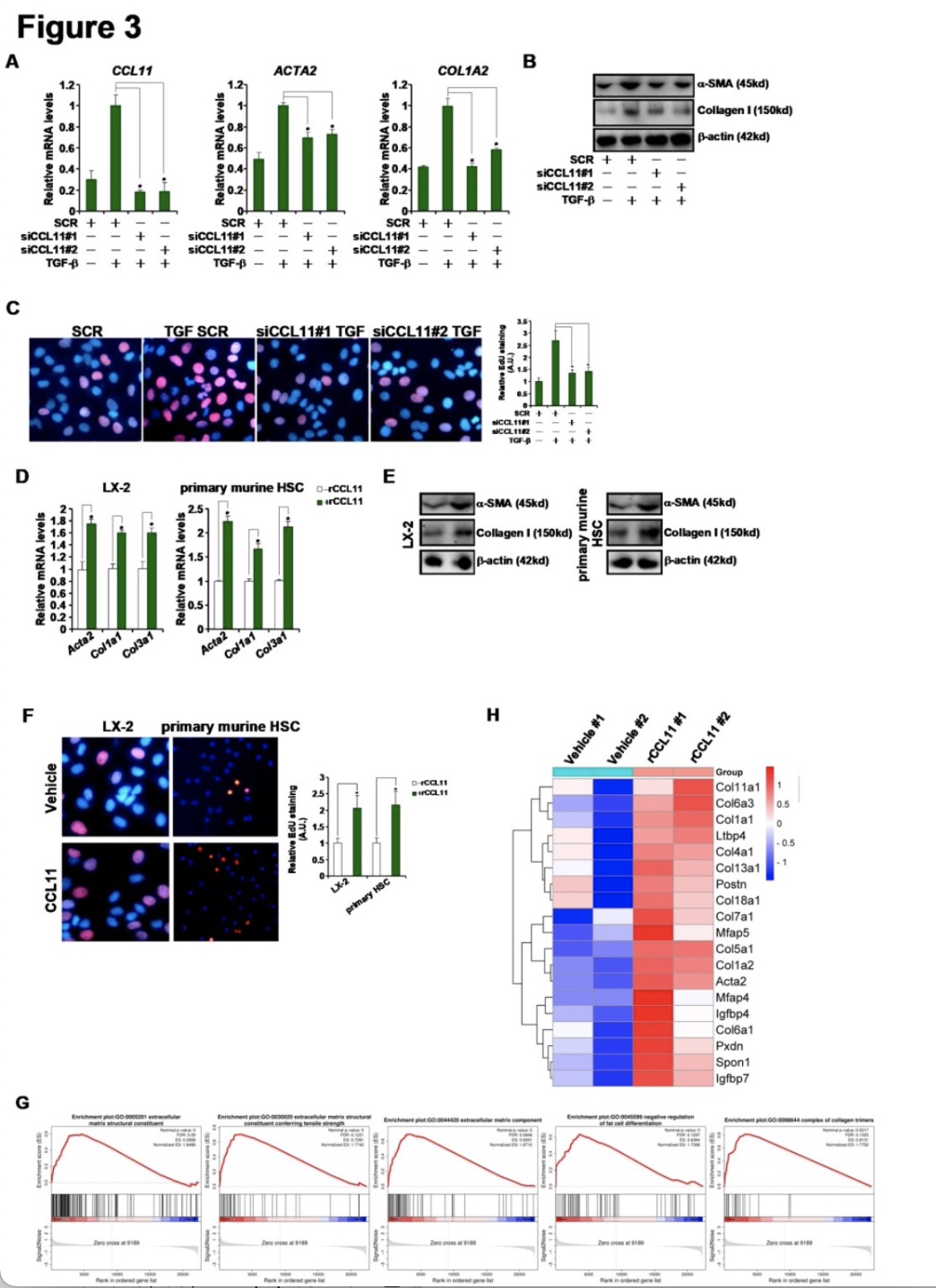
图3 CCL11在体外调节HSC的活化。
(图源:M. Kong, et al, Hepatology,2023)
当从CCL11敲除小鼠(CCL11-/-)和野生型(WT)同窝小鼠中分离出原代HSC,并允许其在体外进行自发激活时,发现CCL11缺失会降低HSCs的激活,这可以从肌成纤维细胞标记基因下调(图4A, 4B)和细胞增殖减慢(图4C)得到证明。接下来,在体内评估CCL11缺乏对肝纤维化的影响。为此,CCL11-/-小鼠和WT小鼠注射CCl4诱导肝纤维化。(图4D和图4E)所示,CCL11-/-小鼠血浆ALT和AST水平明显低于WT小鼠。更重要的是,缺乏CCL11抑制了肝脏中一组促纤维化基因的表达(图4F)。此外,组织学染色在CCL11-/-肝脏中检测到的胶原区域比在WT肝脏中更少(图4G)。CCL11缺失也导致肝脏中F4/80+巨噬细胞、Ly6G+中性粒细胞、ECP+嗜酸性粒细胞浸润明显减少(图4G)。肝羟脯氨酸定量证实,CCL11缺失可减弱小鼠肝纤维化(图4H)。在BDL模型中也有类似的观察结果。
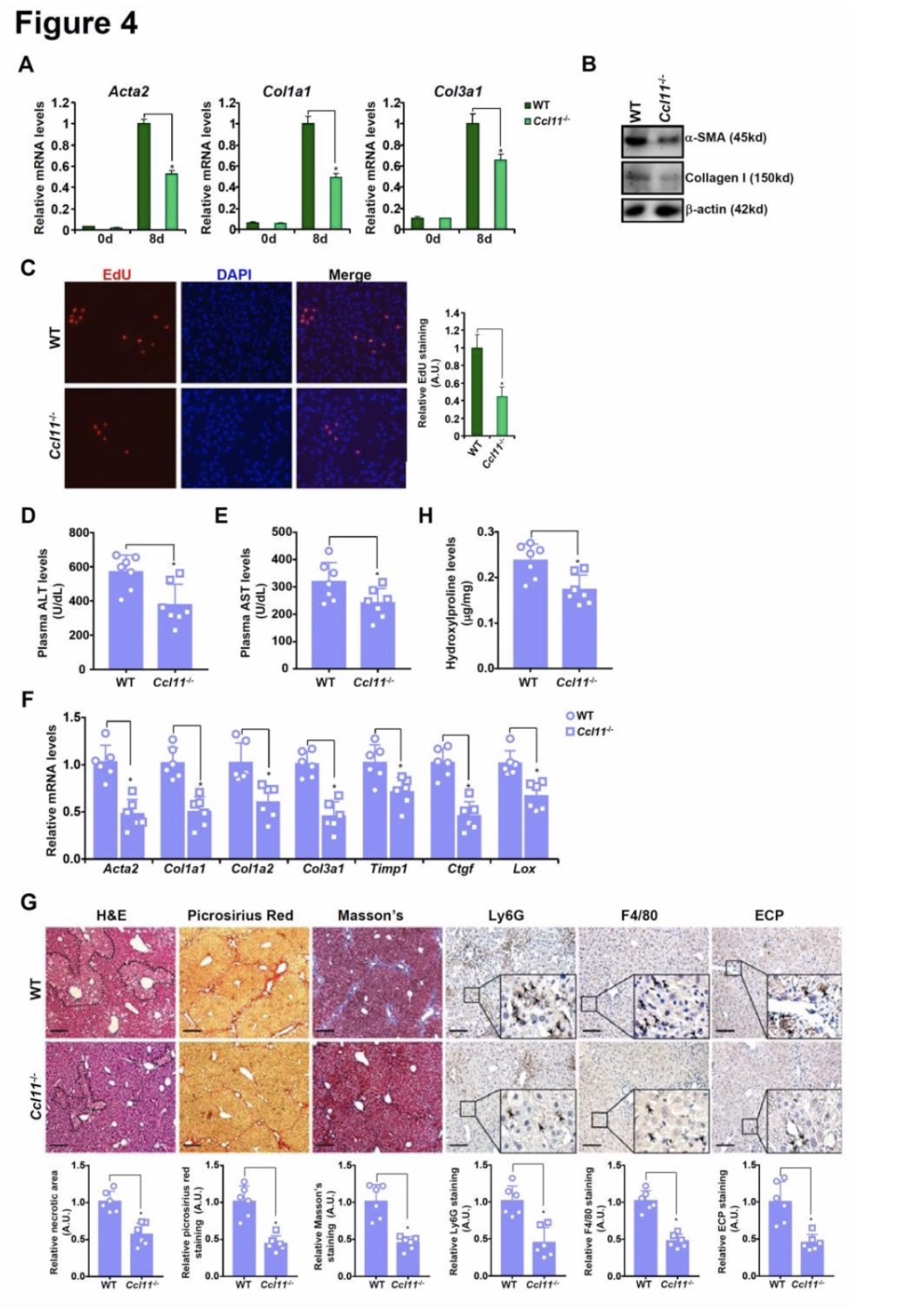
图4 CCL11整体缺乏可减轻小鼠的肝纤维化。
(图源:M. Kong, et al, Hepatology, 2023)
由于CCL11似乎主要来源于肝纤维化过程中的HSCs,因此研究者试图确定HSCs特异性CCL11缺失是否会导致肝纤维化的改善。为此,针对CCL11的shRNA被放置在LRA T启动子/增强子序列的下游,并包装成慢病毒。qPCR分析显示,慢病毒介导的shCCL11传递降低了HSCs中的CCL11水平,但在肝细胞、Kupffer细胞、肝窦内皮细胞或中性粒细胞中没有降低。C57/B6小鼠经尾静脉注射携带shCCL11或shC的慢病毒,然后注射CCl4(图5A)。血浆ALT(图5B)和AST(图5C)水平比较表明,HSCs特异性CCL11缺失并未显著改变BDL诱导的肝细胞坏死。事实上,尽管H&E染色证实了CCL11 -缺失小鼠和对照组小鼠之间的肝细胞坏死没有变化,但天狼猩红染色和马松染色均提示CCL11 -缺失小鼠的肝纤维化较对照小鼠有所减弱(图5D)。qPCR(图5E)和WB(图5F)显示,与对照组相比,CCL11缺失的肝脏中肌成纤维细胞标记基因集体下调。最后,研究发现HSCs特异性CCL11耗竭降低了肝脏羟脯氨酸水平(图5G)。再次,在BDL模型中验证了CCL11敲低对肝纤维化的作用。
为了进一步验证CCL11在肝纤维化中的作用,将CCL11靶向的shRNA放置在Postn启动子的下游,并包装到AAV8 (AA V-shCCL11)中以耗尽成熟肌成纤维细胞中的CCL11。qPCR证实,与AAV-shC相比,AAV- shcl11显著降低了肌成纤维细胞(激活的HSCs)中CCL11的表达,但在肝细胞中没有降低。在CCl4模型和BDL模型中,肌成纤维细胞特异性CCL11缺失显著减弱了肝纤维化,而不改变肝损伤和肝免疫浸润。总的来说,这些数据表明起源于HSCs的CCL11,可能促进肝纤维化。
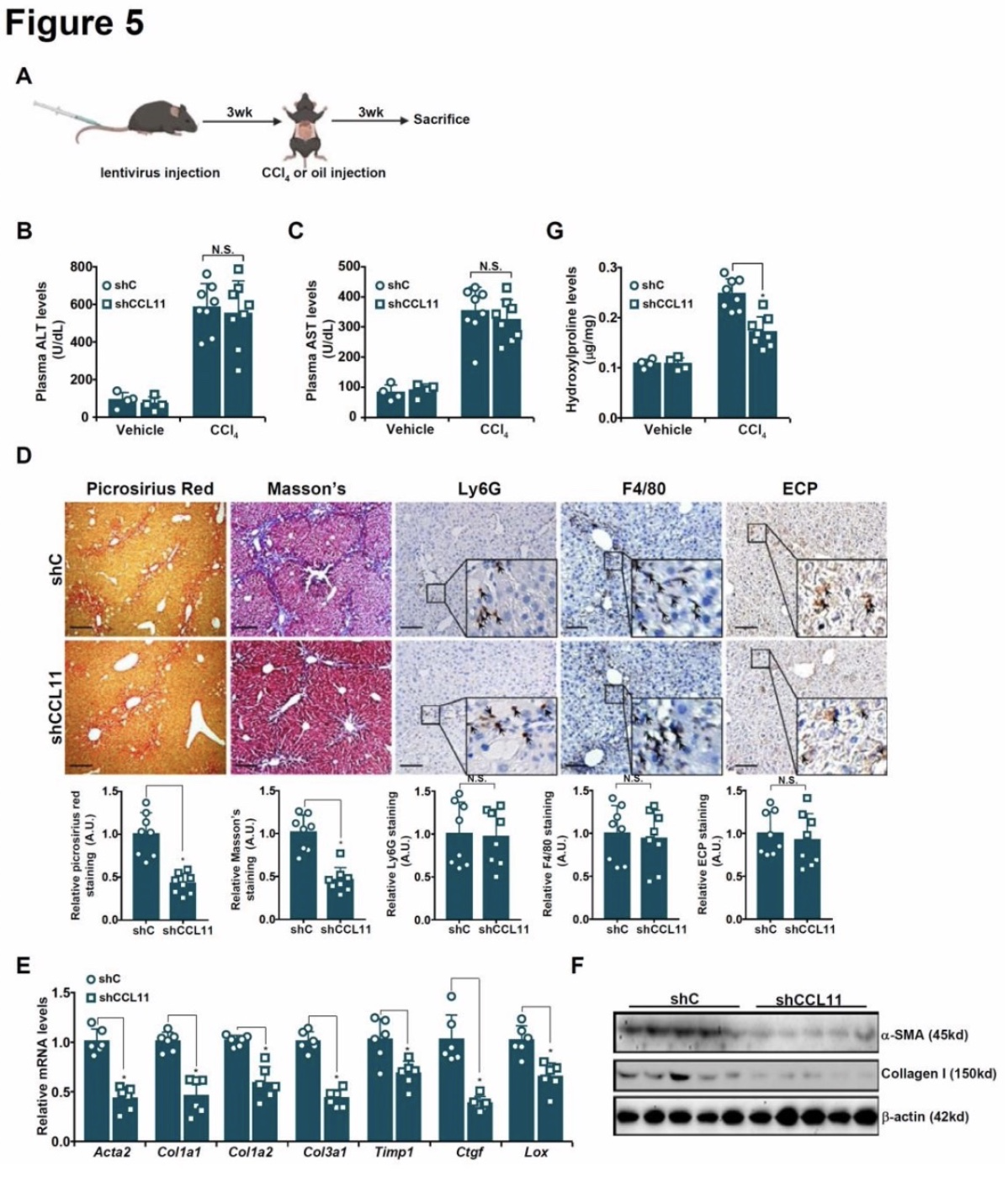
图5 HSC靶向CCL11缺失可缓解小鼠肝纤维化
(图源:M. Kong, et al, Hepatology,2023)
为了解决CCL11可能促进HSC激活和肝纤维化的机制,研究者使用RNA-seq比较了从CCL11 -/-小鼠和WT小鼠分离的HSC的转录组。主成分分析(PCA)表明,CCL11缺失导致细胞转录组发生了巨大变化(图6A)。以p<0.05和1.5Xfold变化为临界值,鉴定出1733个差异表达基因(图6B)。GO分析(图6C)和GESA分析(图6D)均显示CCL11缺失主要影响肌成纤维细胞成熟相关通路。有趣的是,Notch信号通路的配体Jagged 1 (Jag1)已被证明在肝纤维化中发挥作用(29,30),被发现受CCL11缺失的影响最大(按FDR排序,图6E)。重组CCL11处理可上调体外培养的HSCs中Jag1的表达。相反,CCL11缺失或删除(图6F, 6G)会下调Jag1的表达。此外,整体CCL11删除或HSCs特异性CCL11缺失降低了小鼠肝脏中的Jag1水平。
HOMER分析提示几种促纤维化转录因子,包括STA T3、TEAD4和NF- B的活性因CCL11缺失而受到负面影响(图6H)。ChIP检测证实,暴露于CCL11刺激了HSCs中STAT3、TEAD4和NF-κB向Jag1启动子的募集。相反,与WT小鼠相比,从CCL11-/-小鼠分离的HSCs中检测到STA T3、TEAD4和NF-κB的抑制结合(图6I)。
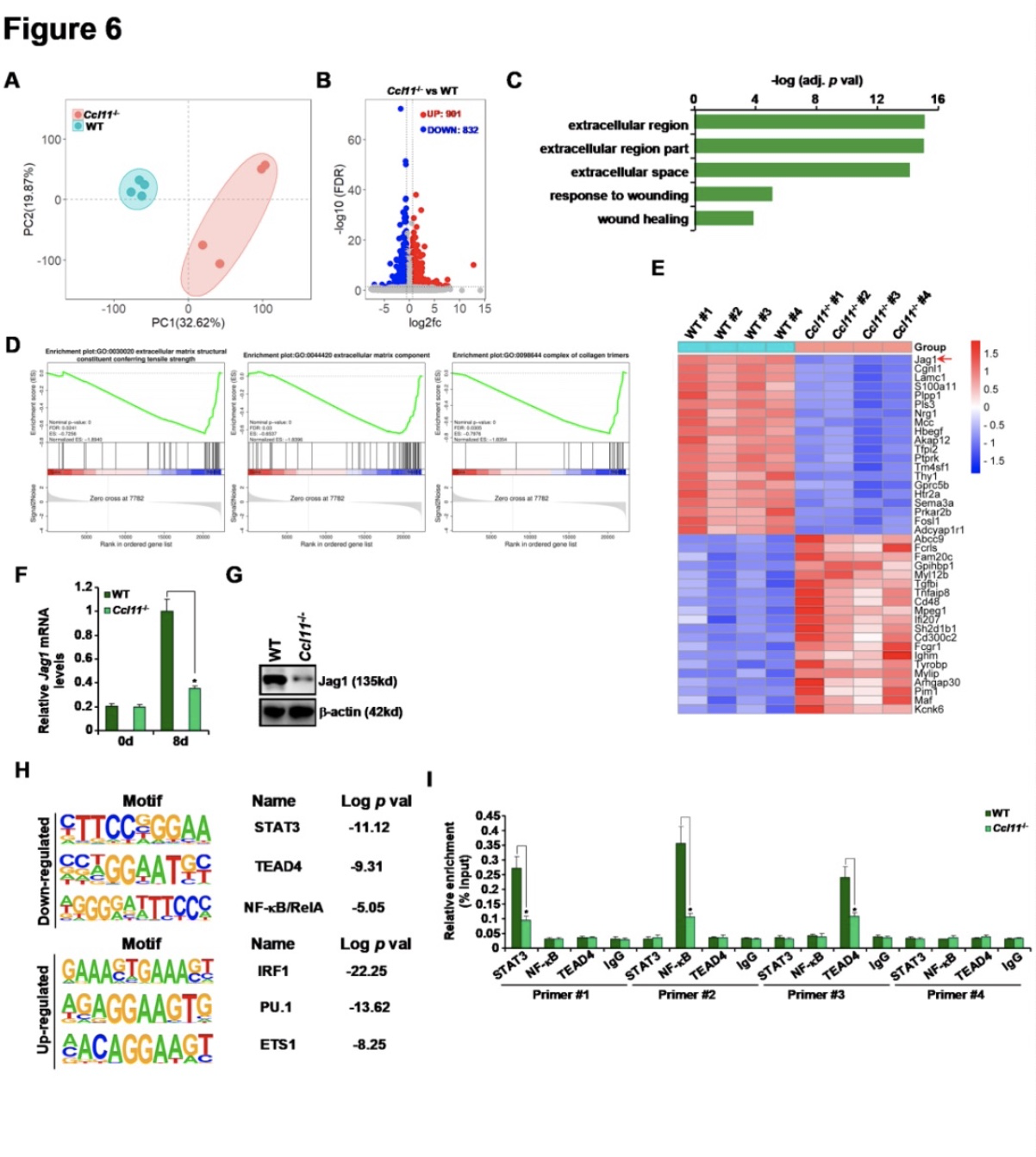
图6 CCL11在HSC激活过程中调节Jag1转录。
(图源:M. Kong, et al, Hepatology,2023)
为了验证CCL11和Jag1之间的功能关系,研究者进行了以下实验。当HSC暴露于重组CCL11时,Jag1的敲除抑制了肌成纤维细胞标记基因的上调。相反,通过腺病毒介导的Jag1载体的传递,Jag1表达的重建使CCL11缺失的HSCs中肌成纤维细胞标记基因的表达正常化,使其水平接近野生型HSCs(图7A, 7B)。值得注意的是,Notch1或Notch 3的缺失完全取消了Jag1过表达对HSCs激活的作用,这表明Jag1可能通过Notch1/3发出信号来发挥其促纤维化作用。
接下来,向CCL11-/-小鼠注射携带Jag1表达载体的AAV8,以验证重新引入Jag1将恢复CCl4注射后的纤维化反应的假设。AAV8-Jag1注射液未显著改变CCL11-/-小鼠血浆ALT (图7C)和AST (图7D)水平。通过肌成纤维细胞标记基因的qPCR测量(图7E)、肝脏胶原组织的组织学染色(图7F)和肝脏羟脯氨酸水平的定量(图7G)判断,Jag1的重建确实使肝纤维化正常化。同样,Jag1的过表达在很大程度上使小鼠肝脏对BDL的纤维化反应正常化。

图7 CCL11通过激活Jag1转录来调节HSC的激活。
(图源:M. Kong, et al, Hepatology,2023)
首先研究者向小鼠注射CCl4,然后注射CCL11中和抗体(图8A)。结果显示CCL11中和抗体改善了肝损伤,减少了肝脏中的免疫细胞浸润(图8B – 8D)。值得注意的是,通过肝胶原组织的组织学染色(图8D)、肌成纤维细胞标记基因的qPCR检测(图8E)以及肝脏羟脯氨酸水平的定量检测(图8F)判断,与注射同型IgG的小鼠相比,注射CCL11中和抗体的小鼠的肝纤维化显著减弱。在BDL模型中,CCL11中和可改善肝损伤,抑制肝免疫浸润,缓和肝纤维化。
CCR3拮抗剂SB297006抑制了TGF-β在HSCs中诱导肌成纤维细胞标记基因。同样,腹腔注射SB297006损伤后软化CCl4诱导和bdl诱导小鼠肝纤维化。最后,CCR3阻断抗体缓解了CCl4模型和BDL模型的肝纤维化。
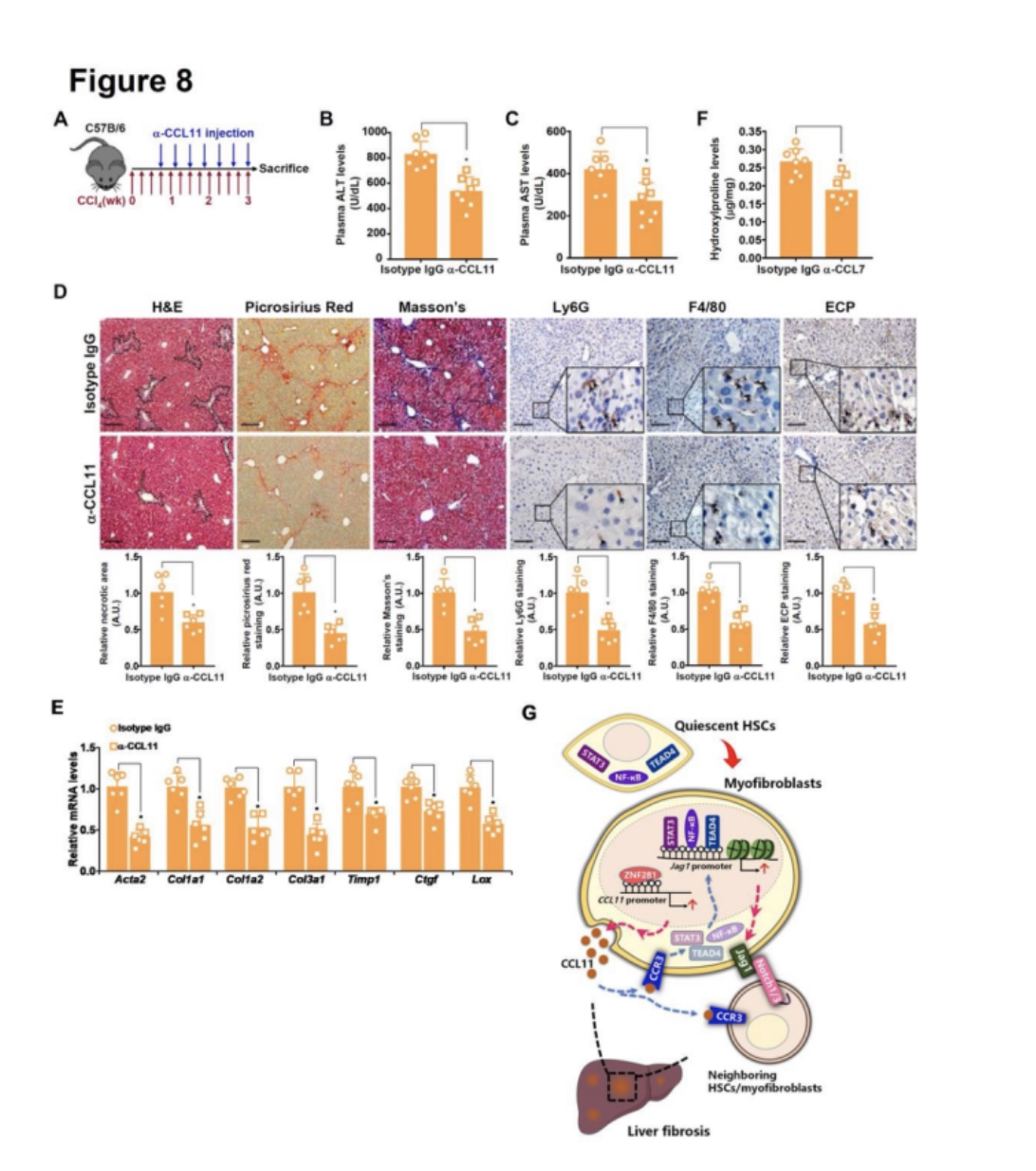
图8 干扰CCL11-CCR3轴可减轻小鼠的肝纤维化。
(图源:M. Kong, et al, Hepatology,2023)
HSCs-肌成纤维细胞转变是肝纤维化的一个标志性事件。研究者在这篇文章中描述了一种新的调节机制,即CCL11,一种以前被认为介导嗜酸性粒细胞转运的趋化因子,它能促进了HSC激活和肝纤维化(图8G)。在肝纤维化过程中,CCL11在HSCs中选择性诱导,但在肝实质细胞中不诱导,其潜在机制目前尚不清楚,有待进一步研究。其次RNA测序数据表明,外源性CCL11刺激会使HSCs倾向于肌成纤维细胞样表型,而CCL11缺失会抑制肌成纤维细胞特征,研究者认为CCL11的促纤维化作用可能是HSCs固有的。值得注意的是,种系CCL11缺失或对CCL11- ccr3级联的整体干扰同时减少了肝损伤和肝纤维化,而HSCs特异性CCL11敲除并没有明显改变肝损伤。这些观察结果清楚地表明,HSCs来源的CCL11可能主要以旁分泌/自分泌的方式促进HSCs-肌成纤维细胞的转变。这个有趣的问题需要在未来使用额外的、谱系特异性的CCL11转基因小鼠品系的研究中加以澄清。通过RNA序列筛选以及转录和功能分析验证,发现Notch信号配体Jag1直接位于CCL11的下游,对于CCL11介导的成纤维反应至关重要。在未来的研究中,利用这些靶点抗体作为肝纤维化模型的治疗方法,肯定会为目前的发现增加更多的转化价值。总之,研究者的数据支持CCL11在介导HSCs -肌成纤维细胞转化和肝纤维化中的作用。鉴于药物干扰CCL11/CCR3信号通路在模型动物中显示出阻断肝纤维化的效果,将CCL11-Jag1轴作为潜在的靶向治疗路径是合理的。
