18101298214
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
首页
实验手册
揭示铁死亡监测全新机制
发布日期:2023/8/14 17:23:00
铁死亡
是一种铁依赖性磷脂过氧化作用驱动的细胞死亡过程,在多种病理状况中起着至关重要的作用,包括癌症、缺血性器官损伤和退行性疾病等[1]。目前研究发现的两种主要铁死亡监测机制:一种是由谷胱甘肽过氧化物酶4 (glutathione peroxidase 4, GPX4)介导的,通过将磷脂过氧化物(phospholipid peroxides, PLOOH)还原为相应的磷脂醇来抑制铁死亡;另一种是由FSP1、DHODH、NOS2和GCH1等酶介导的,这些酶会产生具有自由基捕获抗氧化(antioxidant, RTA)活性的代谢物,防止PL过氧化,抑制铁死亡[1-3]。
肿瘤细胞铁死亡如何参与或抑制抗肿瘤免疫一直存在争议, 确定是否存在独立于GPX4和RTA的额外监视机制尤为重要,可为开发新型联合疗法提供新靶点
。
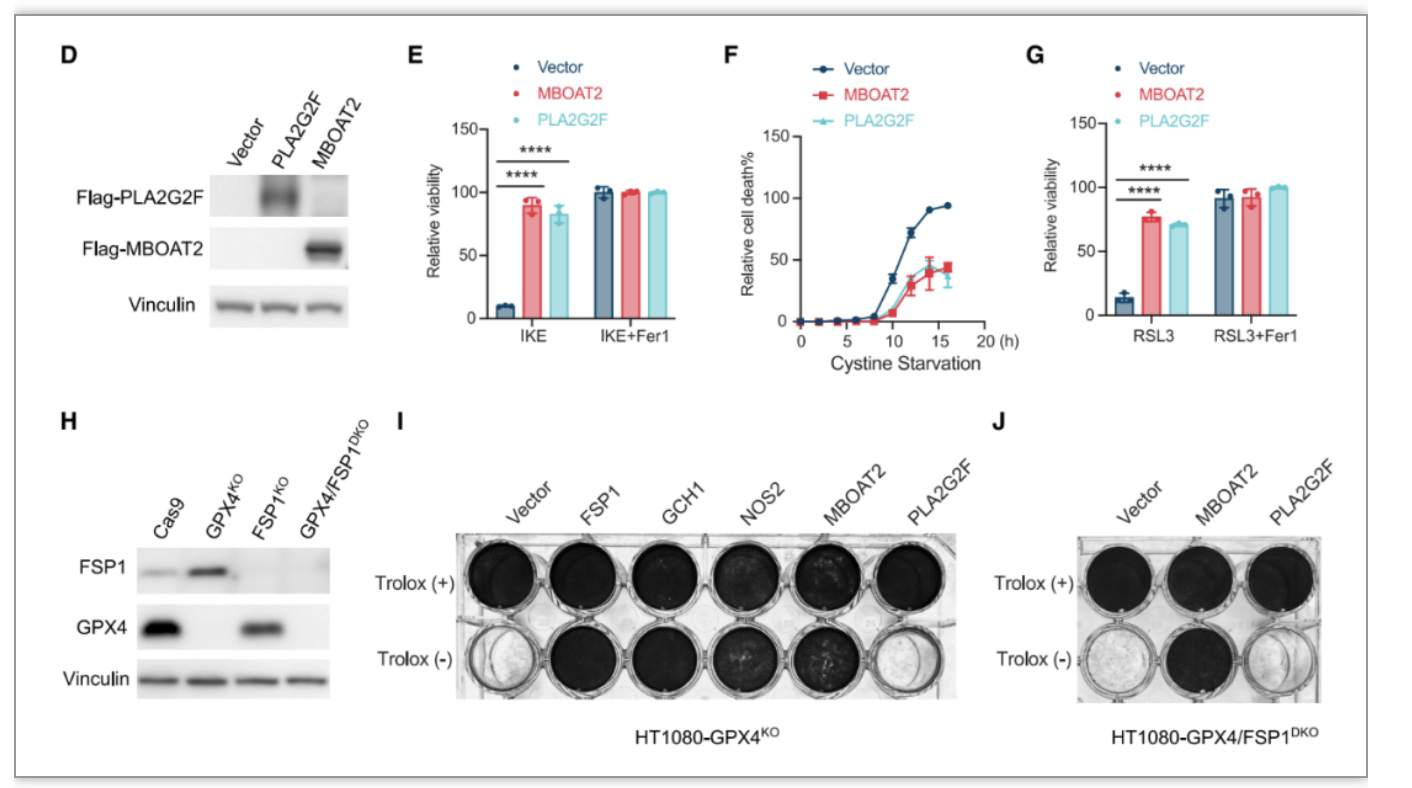
作者使用GPX4抑制剂RSL3和胱氨酸饥饿诱导人纤维肉瘤HT1080细胞铁死亡,进行CPISPR激活筛选,发现7个基因与抵抗铁死亡相关的基因,包括5个已确定的铁死亡抑制因子:SLC7A11、NFE2L2/NRF2、FSP1、GCH1和NOS2,还有两个未被报道具有铁死亡调控功能的脂质修饰酶MBOAT2和PLA2G2F。为了验证MBOAT2和PLA2G2F是否具有抑制铁死亡的功能,作者通过在HT1080细胞中过表达MBOAT2和PLA2G2F,再给予铁死亡诱导剂处理。作者发现分别过表达MBOAT2和PLA2G2F均能显著抑制RSL3或者Erastin诱导的铁死亡。
在敲除GPX4,或者同时敲除GPX4和FSP1的HT1080细胞中,过表达MBOAT2仍然能显著抑制人成纤维肉瘤细胞细胞铁死亡,表明MBOAT2是一种不依赖于GPX4和FSP1的强效铁死亡抑制基因。
图1. MBOAT2是一种不依赖于GPX4/FSP1的铁死亡抑制因子
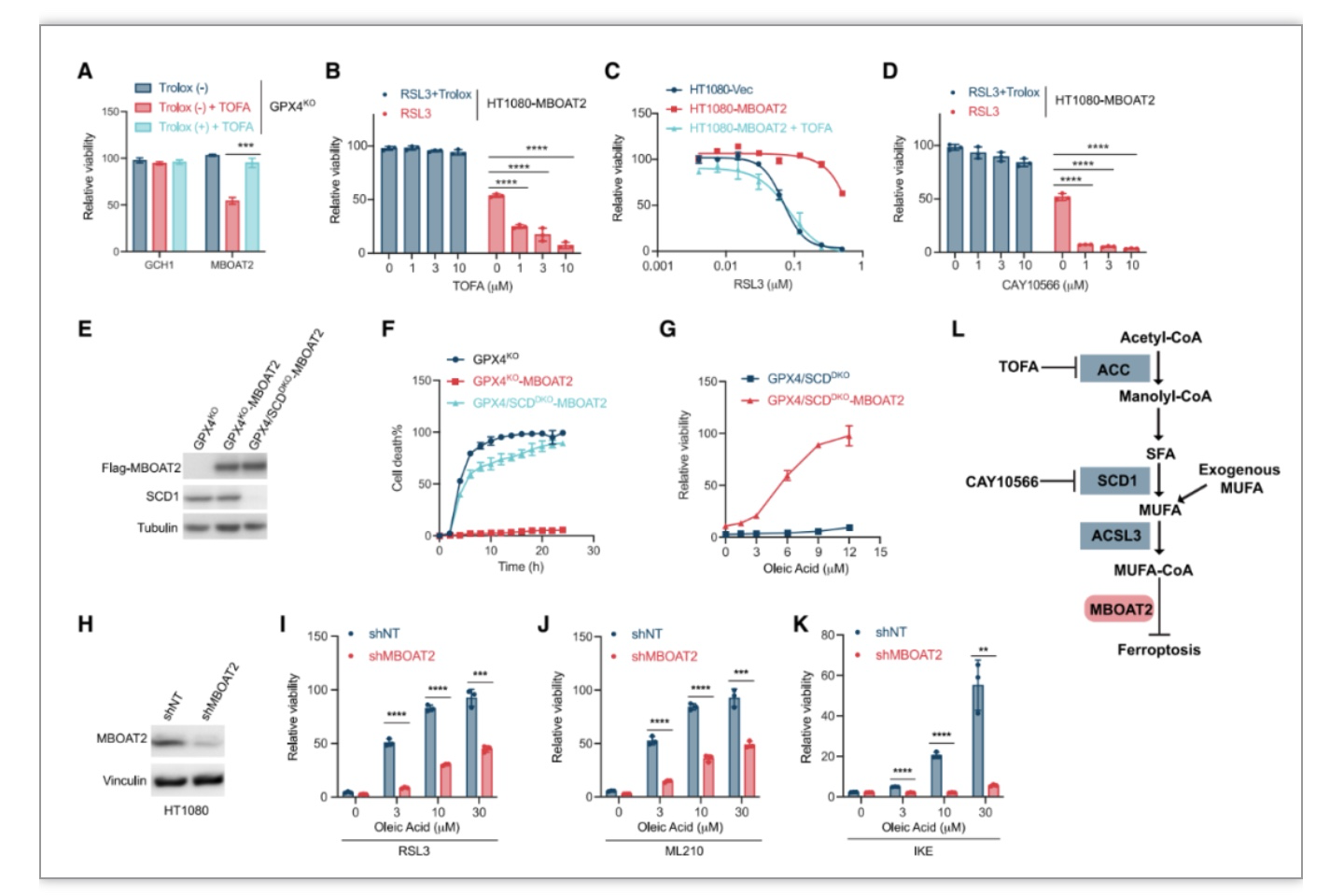
作为膜结合O-酰基转移酶(Membrane-bound O-acyltransferases, MBOAT)家族的成员,MBOAT2具有溶血磷脂酰基转移酶(LPLAT)活性,偏好将单不饱和脂肪酸(MUFA)转移到磷脂酰乙醇胺(Lyso-PE),也可能转移到溶血磷脂酰胆碱 (Lyso-PC) 和溶血磷脂酸(Lyso-PA)[5-6]。MBOAT家族的另一个成员MBOAT5通过将PUFA(多不饱和脂肪酸)转移到PL以提高 PL-PUFA的水平来促进铁死亡。作者推测MBOAT2是否催化MUFA到PL,竞争性降低PL-PUFA含量并最终导致铁死亡抗性细胞状态。MUFA的从头脂肪生成始于乙酰辅酶A羧化酶(ACC)形成丙二酰辅酶A。ACC抑制剂TOFA以剂量依赖性方式诱导MBOAT2过表达的HT1080-GPX4
KO
细胞以及RSL3诱导过表达MBOAT2的HT1080细胞铁死亡,并且可以完全消除MBOAT2的铁死亡抑制活性,表明MBOAT2铁死亡抑制作用依赖于脂肪生成。从头合成的饱和脂肪酸(SFA)通过SCD1转化为MUFA,作者发现使用SCD1抑制剂CAY10566或者敲除SCD1消除了MBOAT2对RSL3或GPX4敲除诱导的铁死亡的保护活性。并且MBOAT2过表达后,
外源MUFA的保护功能在HT1080-GPX4/SCD1
DKO
细胞中进一步增加,而MBOAT2敲低显着减少了外源性MUFA的保护作用,以上数据表明MBOAT2的铁死亡抑制功能需要内源性或外源性MUFA。
图2. MBOAT2 的铁死亡抑制功能需要内源性或外源性MUFA
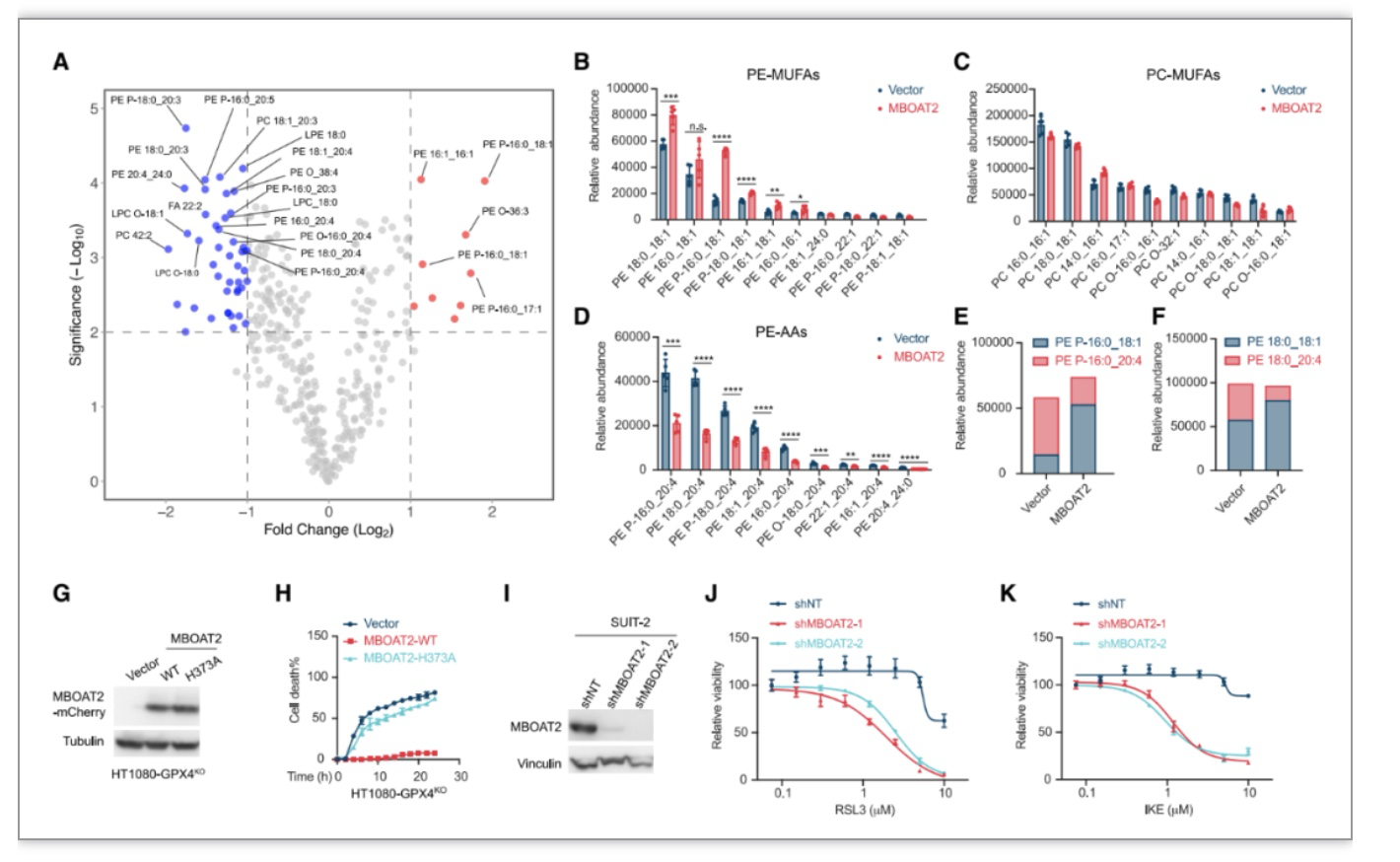
由于MBOAT2依赖MUFA抑制铁死亡,作者猜想MBOAT2是否通过PL重塑抑制铁死亡。作者使用HT1080和MBOAT2过表达的HT1080细胞进行非靶向脂质组学分析,
发现MBOAT2过表达选择性增加含有单不饱和脂肪酸的磷脂酰乙醇胺(PE-MUFA)的生成,降低含有多不饱和脂肪酸的磷脂酰乙醇胺(PE-PUFA)的生成。
作者推测存在竞争性PE重塑模型,MBOAT2 选择性地将MUFA-CoA转移到lyso-PE以竞争性减少PUFA-CoA掺入lyso-PE。组学数据进一步分析发现与对照相比,MBOAT2的过表达选择性地增加PE-MUFAs, PC-MUFA和其它PL-MUFA并未受到显着影响,而PE-PUFA 在MBOAT过表达后显着降低。
PE-PUFA是磷脂过氧化的首选底物和铁死亡敏感性的主要决定因素,MUFA偏好lyso-PE酰基转移酶MBOAT2与 PUFA偏好的lyso-PE酰基转移酶在lyso-PE竞争,在具有主要 MBOAT2活性的特定细胞或组织中,PE重塑促使细胞转向高PE-MUFA水平但低PE-PUFA 水平的状态,导致铁死亡抗性。
图3. MBOAT2通过磷脂重塑抑制铁死亡
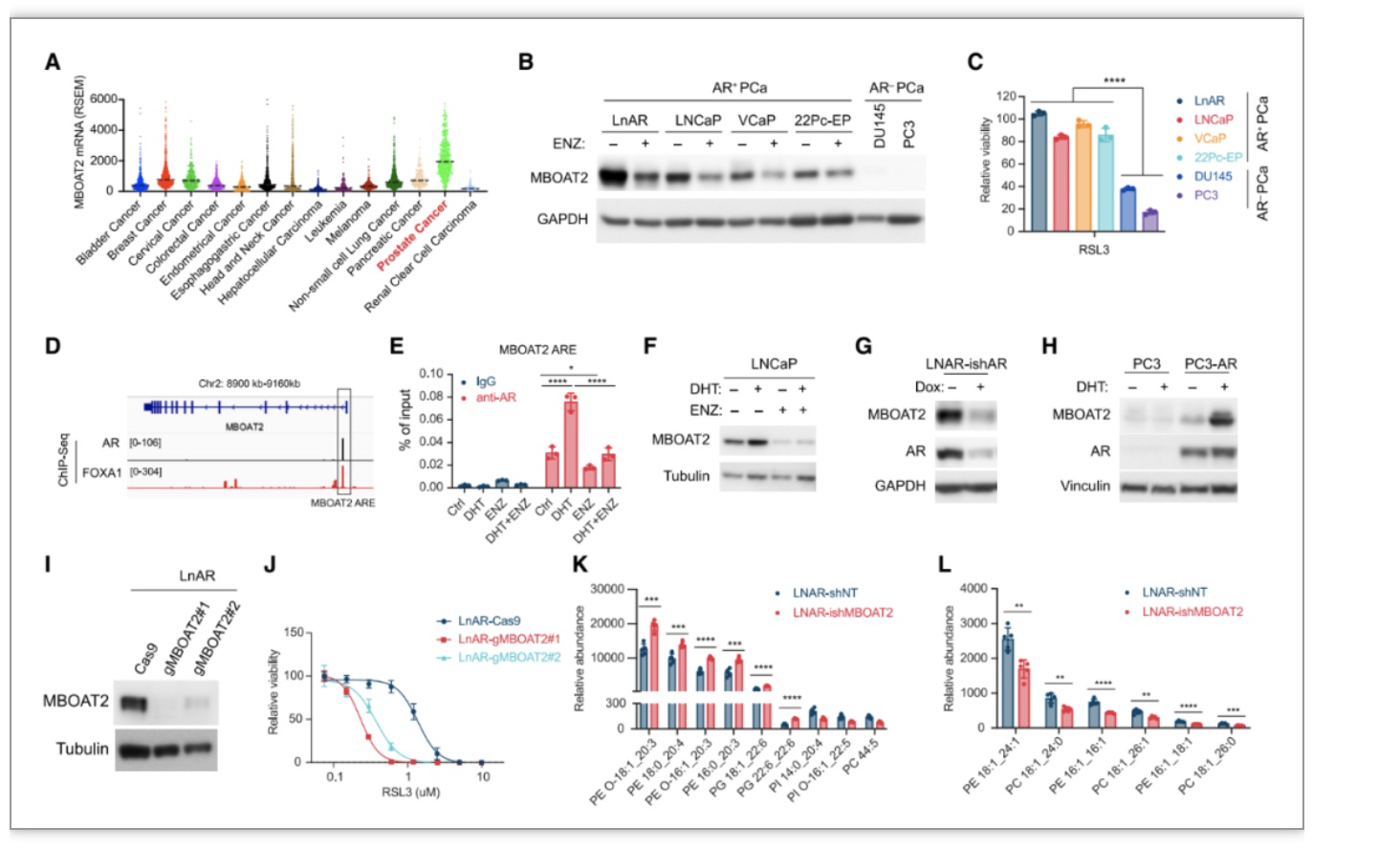
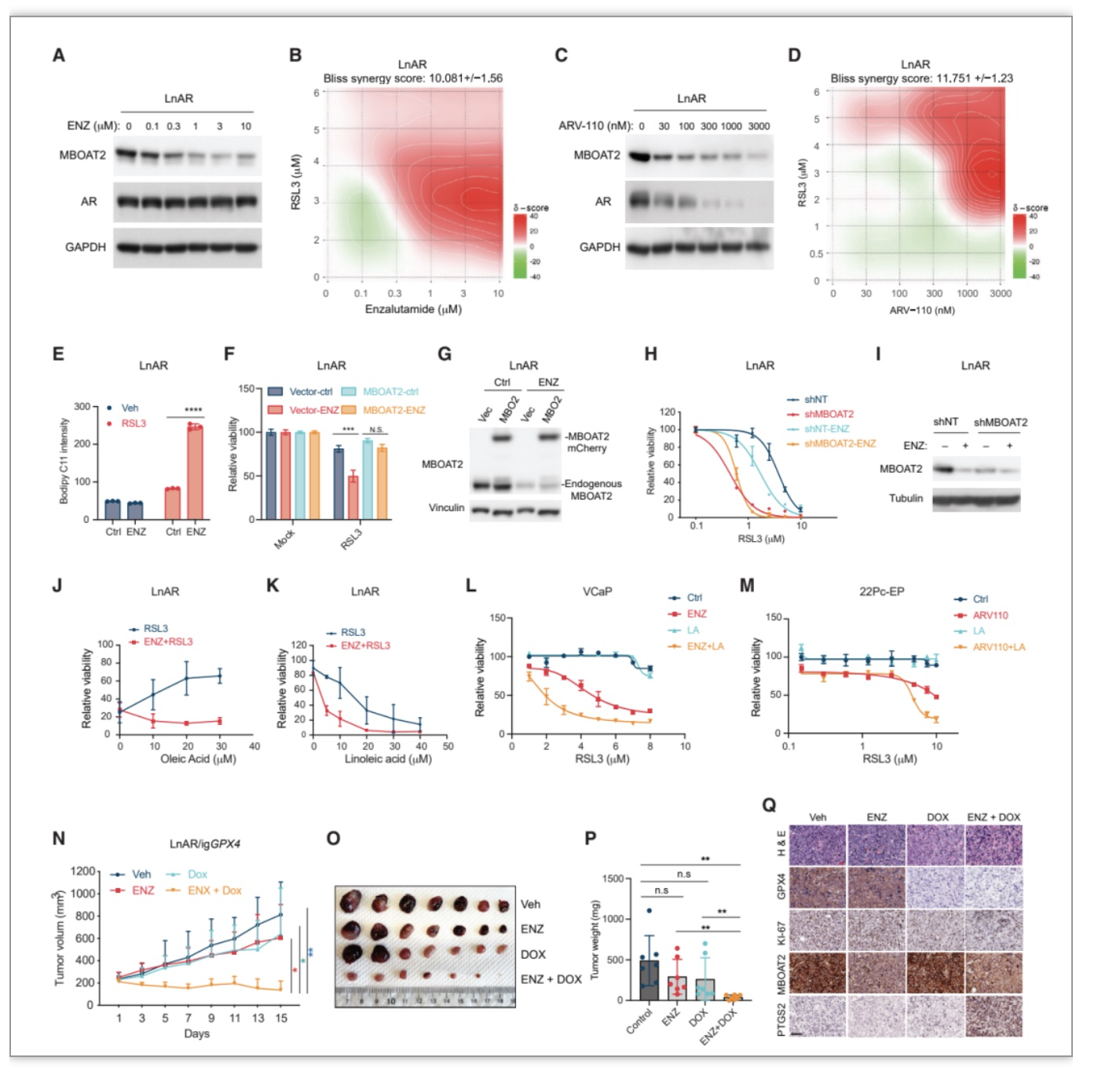
作者通过数据库比较MBOAT2 mRNA水平发现MBOAT2在前列腺癌(PCa)中特异性上调。MBOAT2 mRNA与人类PCa样本中的雄激素受体(AR) mRNA呈正相关,在AR
+
PCa细胞系中高表达,在AR
-
PCa细胞系中低表达,并且AR
+
PCa细胞系比AR
-
PCa细胞系更能抵抗铁死亡。通过ChIP-qPCR分析,作者进一步确认AR与MBOAT2 的顺式作用元件结合,并且AR激动剂二氢睾酮(DHT)可促进二者结合,AR拮抗剂ENZ抑制二者结合,并且DHT促进AR
+
PC细胞系中MBOAT2表达,而ENZ降低MBOAT2 表达。作者随后也确认了MBOAT2是否调节AR
+
PCa细胞的铁死亡敏感性。MBOAT2 敲除或敲低使前列腺癌细胞LnAR和LNCaP对RSL3诱导的铁死亡着更敏感。并且MBOAT2敲低通过显着增加PE-PUFA的含量但降低PE-MUFA 的含量来重塑PL。因此,
AR信号通过上调MBOAT2表达并重塑细胞PL,抑制铁死亡。
图4. AR信号通过MBOAT2调节前列腺癌细胞的铁死亡敏感性
AR
+
PCa细胞中MBOAT2高表达促进了铁死亡抗性,作者猜想拮抗AR作用是否会通过下调MBOAT2促使肿瘤细胞对铁死亡诱导敏感,从而成为一种潜在的治疗策略?作者发现ENZ (FDA批准的第二代针对AR靶向治疗剂)与ARV-110(一种临床阶段的AR降解剂)均以剂量依赖性方式显著抑制内源MBOAT2表达,并且使多种AR
+
PCa细胞对RSL3 诱导的铁死亡敏感。多西环素(Dox)可诱导Cas9表达,作者使Dox处理LnAR-iCas9-gGPX4 (LnAR-igGPX4)细胞系诱导GPX4敲除,发现ENZ可进一步增强细胞中GPX4敲除引发的铁死亡。在LnAR-igGPX4移植瘤动物模型中,
Dox或ENZ单独处理仅适度抑制肿瘤生长,而Dox与ENZ的组合可显著抑制肿瘤生长,表明ENZ与铁死亡诱导的联合治疗是治疗AR
+
PCa的一种潜在方案。
图5. AR拮抗剂使AR
+
前列腺癌细胞对铁死亡敏感
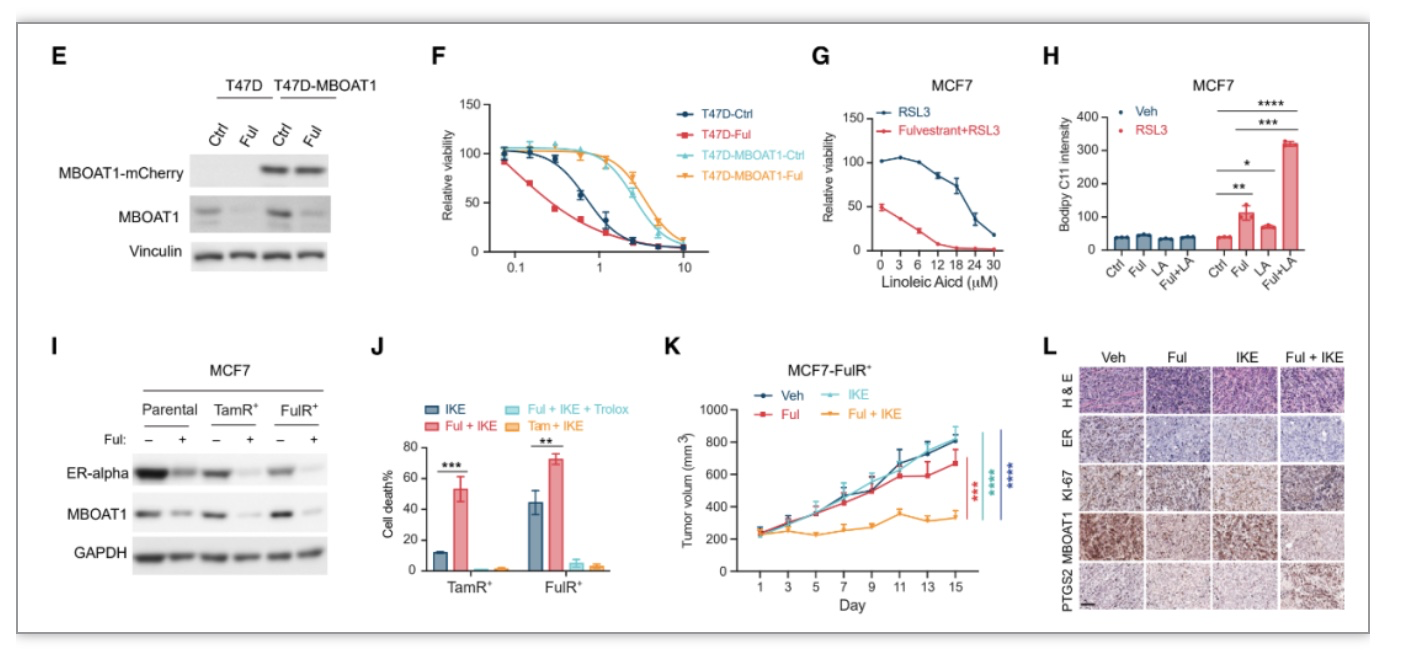
另外作者发现MBOAT家族中除了MBOAT2,MBOAT1也可以抑制GPX敲除或者RSL3诱导的铁死亡。与MBOAT2一样,MBOAT1通PL重塑抑制铁死亡,并偏向于PE-MUFA。有趣的是,
与MBOAT2相反, MBOAT1在女性癌症中高表达,包括卵巢癌、乳腺癌和子宫内膜癌等
。
与MBOAT2功能发现类似,
作者发现雌激素(ER)受体信号调控MBOAT1表达,MBOAT1表达下调会导致ER
+
乳腺癌细胞T47D和MCF7对铁死亡更敏感。
ER抑制剂Fulvestrant与铁死亡诱导剂IKE单独使用不能抑制ER
+
乳腺癌细生长,相反Ful和IKE的组合显著抑制了肿瘤生长,
提示靶向ER与铁死亡诱导的联合治疗可能是耐药性ER
+
乳腺癌的潜在治疗方法。
图6. ER拮抗剂Fulvestrant通过下调MBOAT1表达促进ER
+
乳腺癌细胞铁死亡敏感性
文章结论与讨论,启发与展望
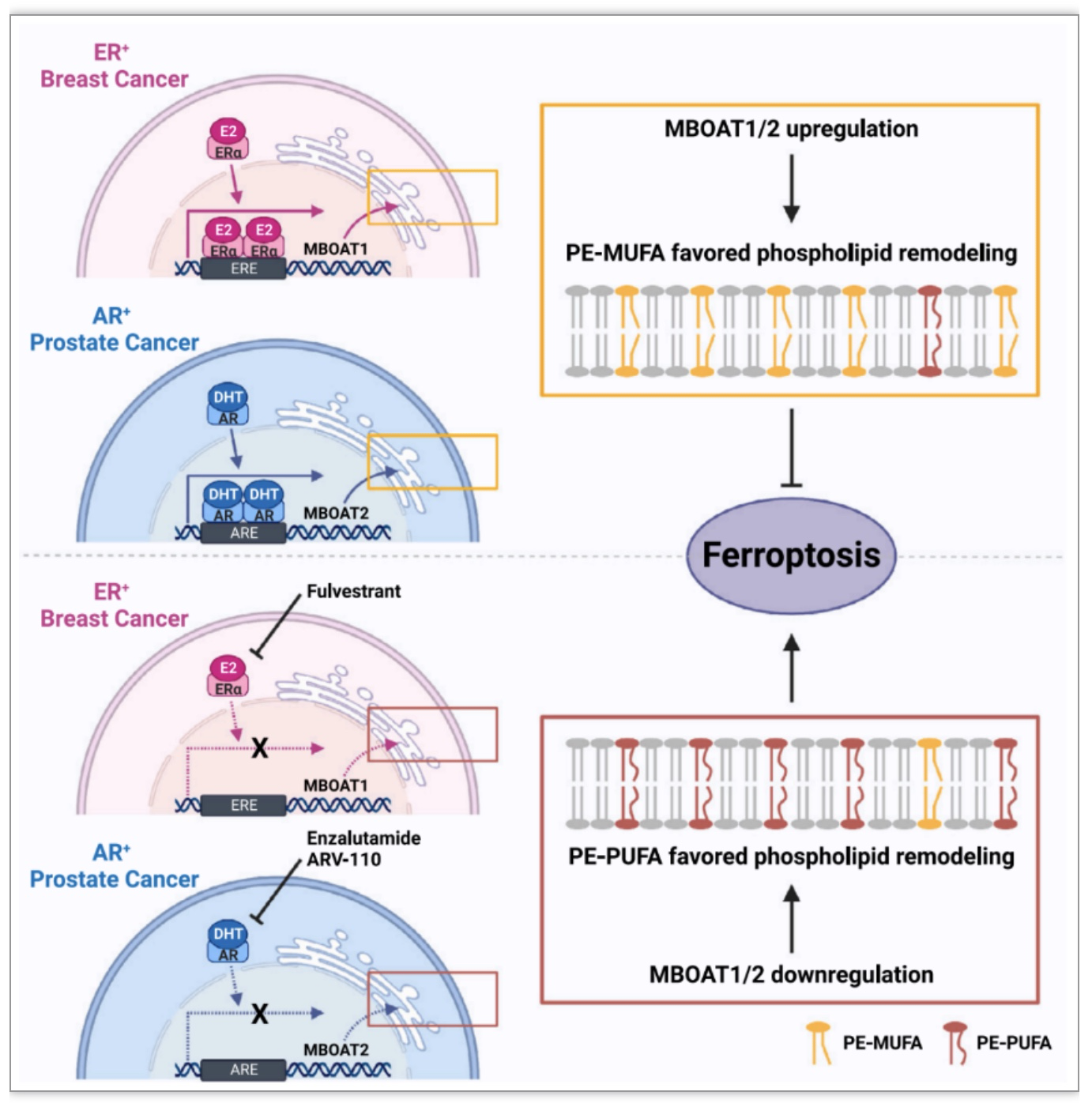
综上所述,该研究阐述了一种调控铁死亡监测的新分子机制,即MBOAT1/2介导的磷脂重塑抑制癌细胞铁死亡,并且MBOAT1和MBOAT2在ER
+
和AR
+
肿瘤中分别受到不同的性激素调节以抑制铁死亡,为癌症联合疗法提供了新见解。然而,减少PL过氧化底物是否是 MBOAT1/2介导的铁死亡抑制的唯一机制仍有待进一步实验确定,以及PL-MUFA抑制铁死亡的机制也有待进一步研究。目前的研究虽然为两种潜在的癌症联合疗法提供了见解,然而到目前为止尚未开发出用于临床的铁死亡诱导剂,从而限制了临床前阶段的研究。
上一篇:
揭示了肿瘤细胞在空间限制性迁移中维持细胞存活并促进转移的分子机制
下一篇:
优化生物反应器生产、加速生物药开发的实用工具
已经到最底了
技术支持:
库价化学
Copyright © 2024北京螽斯羽生物有限公司 备案号:
京ICP备2023018288号-1