18101298214
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
公司首页
实验手册
小分子化合物
细胞培养和检测
定制服务
文献资料
联系我们
订购指南
首页
实验手册
提出A-to-I RNA编辑位点研究新方案
发布日期:2023/8/10 10:15:00
RNA编辑
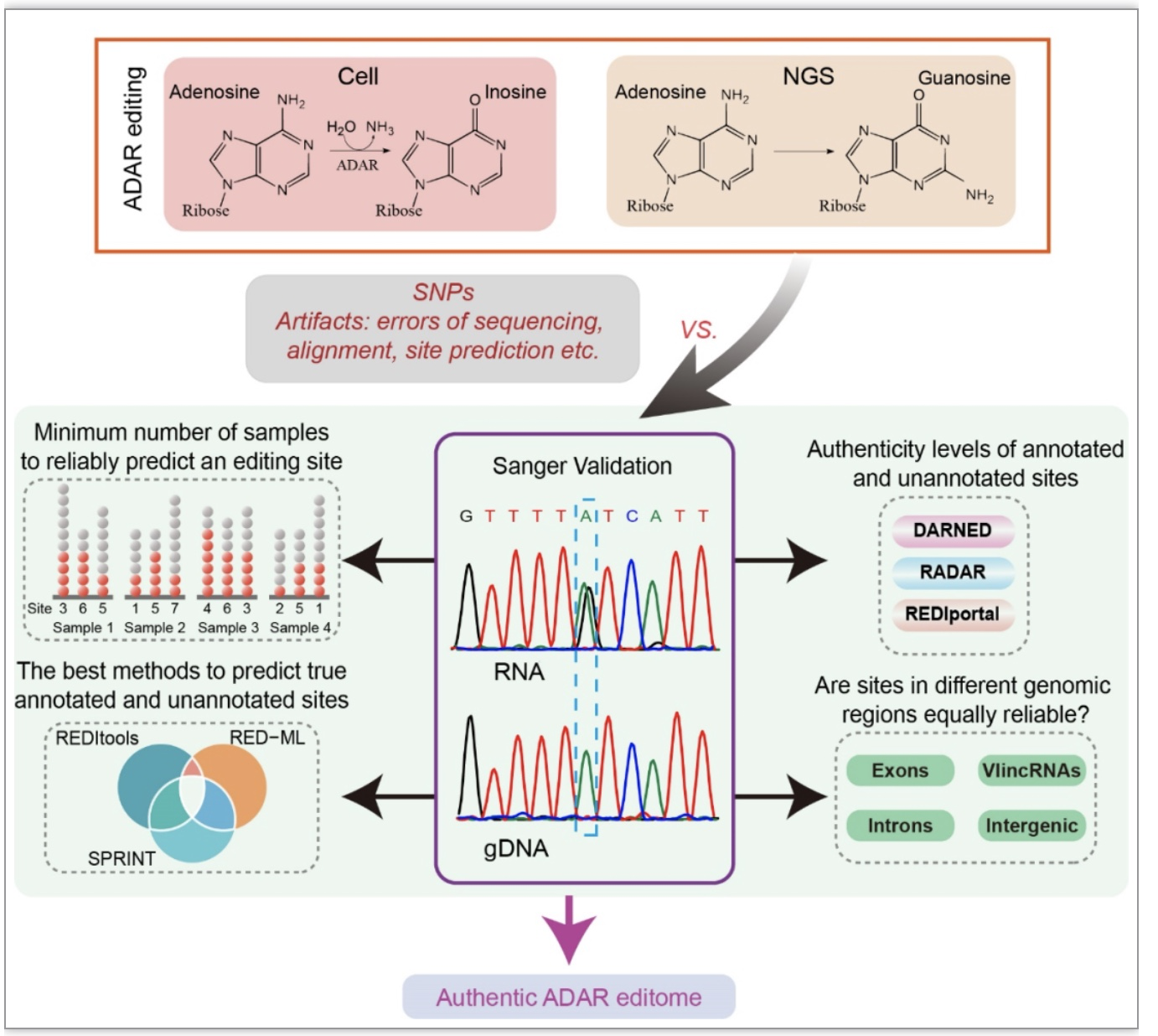
是指在RNA发生改变而DNA保持不变的分子生物学过程[1],是一种转录后修饰,通常包括RNA分子内核苷酸的插入,缺失和碱基转化。细胞内酶催化的腺嘌呤(A)到次黄嘌呤(I)的编辑代表了一类常见的与疾病发生发展相关的转录后RNA编辑。发生在特定位点的A-to-I的转化会导致蛋白质多样性的增加以及[2],mRNA稳定性[3]、可变剪切和表达调控[4]的变化。目前已公布的RNA编辑位点数据库数量庞大,但是其正确性仍有待研究(图1)。
本研究为解决当前
A-to-I
位点预测的高假阳性问题提供了一个潜在解决方案,并基于大量的
Sanger
实验验证公布了一个可靠且全面的人类癌症细胞系
K562
中
A-to-I
编辑位点的列表。
该研究表明大部分真实的人类癌症细胞系的A-to-I编辑位点位于“RNA暗物质”非编码区域,许多发生在人类mRNA的外显子区域的A-to-I编辑位点都是假阳性位点,因此对发生在该区域的解释需格外谨慎。
图1. 研究方案
(图源:Wang,
et al.
,
BMC biol
, 2023)
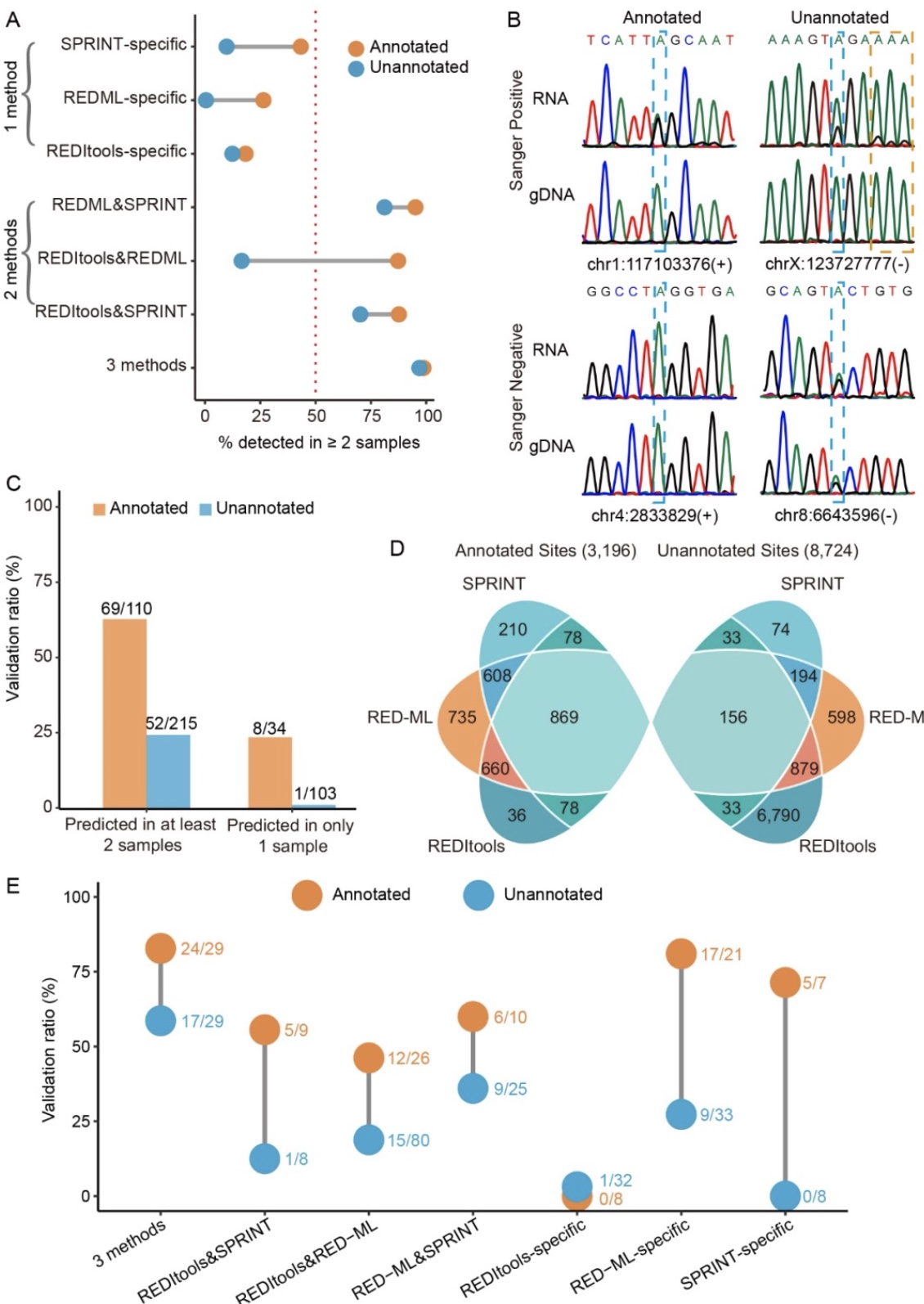
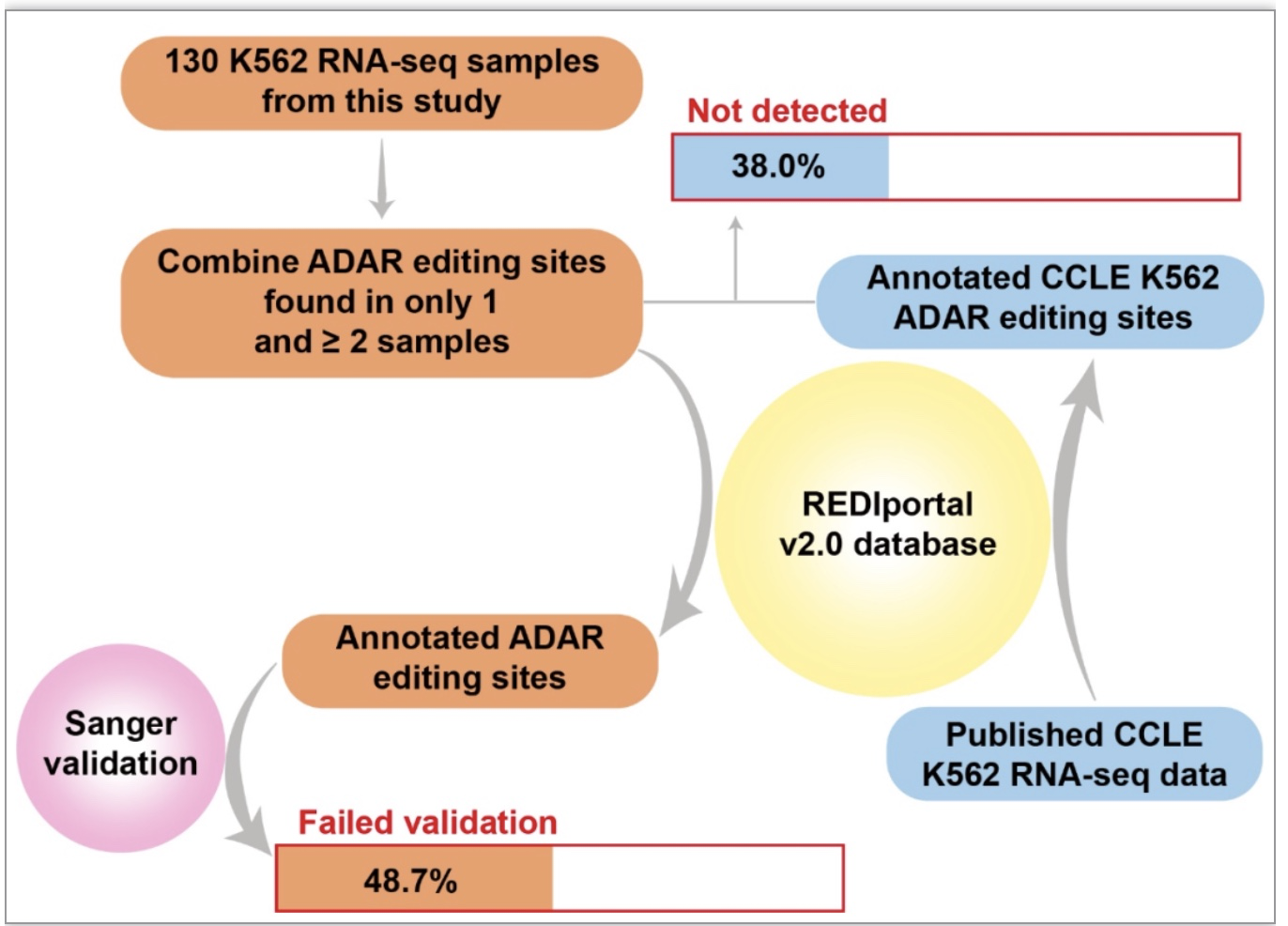
研究分析了114个不同抗癌药物处理不同时间的K562细胞和16个不同慢病毒载体转染的K562细胞共计130个样品的测序数据。每一个样本都由3个不同的分析方法:REDItools、RED-ML、SPRINT进行RNA编辑位点预测。每个样本预测出的所有RNA编辑位点去除人类dbSNP 151和K562基因组的SNP,只保留编辑水平大于0.2的候选位点,得到近20万个位于非重复区域的候选位点。使用三个数据库DARNED, RADAR, REDIportal v2.0已注释位点的合集将候选位点分为已注释和新发现位点。已注释和新发现位点在被检测到的样本数量上有显著差异,大部分只在一个样本中检测到的编辑位点也只被一种分析方法检测到,而在两个及两个以上样本中被检测到的点也具有被至少两种方法检测到的趋势(图 2a-d)。
图2. 真实的A-to-I编辑位点在相同细胞类型的独立RNA测序样本中是可重复的
(图源:Wang,
et al.
,
BMC biol
, 2023)
不同分析方法在检测RNA编辑位点时的表现具有很大差异。在已注释的RNA编辑位点方面,3个方法的主要差异在于灵敏度,而在准确性上差异较小。在新发现的位点中,灵敏度和准确性上这些方法都表现出了不同的结果。多个方法的联合检测能够显著提升准确性,代价是在灵敏度上做了很大牺牲(图 2e)。总之,同时考虑灵敏度和准确性时,即使在使用超过一个方法时仍没有得到令人满意的结果。因此,作者基于候选位点的基因组定位开发了更多的筛选机制来去除假阳性位点。
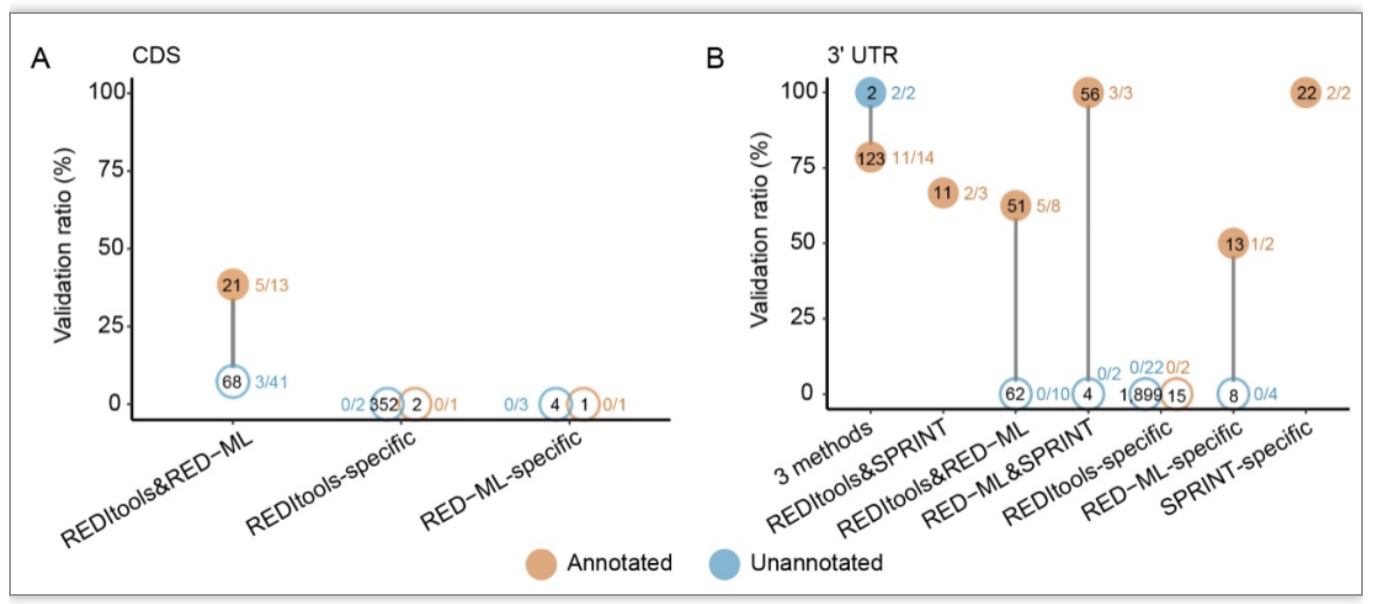
研究发现,新发现RNA编辑位点在外显子区域大量存在,但被测试的86个外显子新发现位点中,仅有5个位点成功验证,其中3个在CDS中,2个在3' UTR(图 3)。而已注释位点在CDS和3' UTR区域的加权验证率显著高于新发现位点。这些结果表明,
在解释外显子中发现的新发现的人类ADAR编辑位点时必须非常小心,因为这些区域的假阳性率可能非常高。即使在已注释的位点中,位于外显子区域的位点也需要进行验证,尤其是位于CDS区域的位点,其验证率远低于其他的已注释RNA编辑位点。
图3. 真实的新发现A-to-I编辑位点在外显子区域很稀少
(图源:Wang,
et al.
,
BMC biol
, 2023)
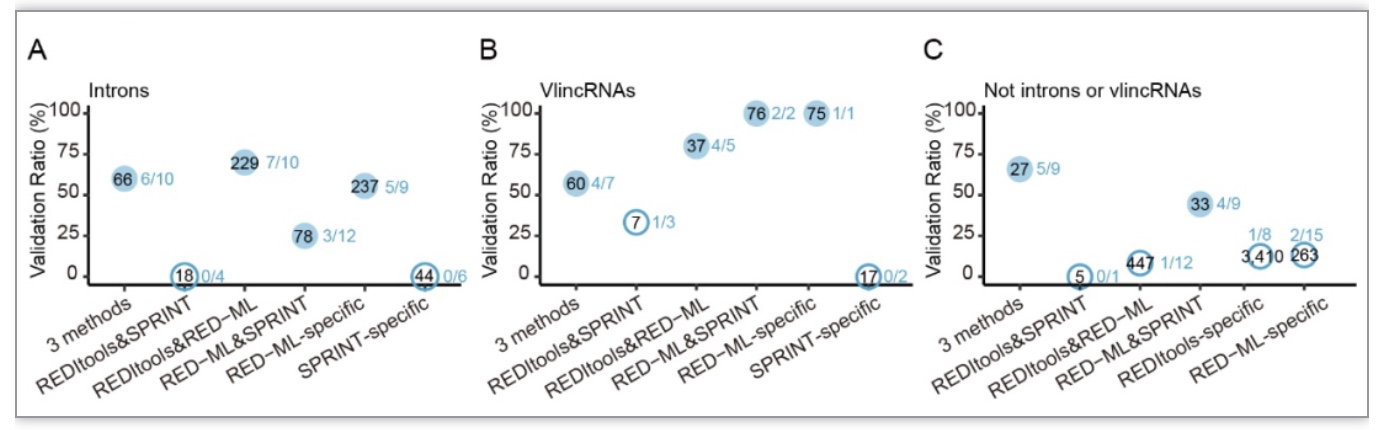
此外,作者还研究了外显子区域以外的RNA编辑位点,发现真实的新发现编辑位点常见于非编码转录本中,尤其是内含子和vlincRNA中。余下的位于非编码基因组区域的新发现位点的验证率都很低(图 4)。
图4. 非编码区域的新发现A-to-I编辑位点的验证比率
(图源:Wang,
et al.
,
BMC biol
, 2023)
上述的结果表明,通过基因组位置和检测方法对新发现位点进行过滤后,生成了一个验证率较高的真实新发现位点列表,图 5a显示了基于上述结果开发的流程。总计发现了989个新发现的位点,其加权验证率为73.5%(图 5c)。由于新发现位点的选择受基因组区域的影响,作者分别分析了已注释和新发现的编辑组的特性,首先,具有增加蛋白质组多样性潜力的ADAR编辑位点在人类癌细胞中非常罕见,在该研究中一共预测了34个高置信度的位于K562细胞的CDS区域的RNA编辑位点(图 5b,c),其中通过Sanger直接验证的只有5个基因中的9个位点(5个已注释和4个新发现位点),其中6个可以引起氨基酸变化。基于已注释编辑位点的结果,位于CDS区域的点占K562细胞系中所有非重复区域编辑位点的比例不超过1%(图 6b)。其次,位于3' UTR区域的位点更为常见,预计它们占人类癌细胞非重复区域编辑位点的约10%(图 5b)且RNA编辑位点在3' UTR富集,但在5' UTR区域的富集则略低(图 5d)。大部分已注释(85.9%)和新发现(97.7%)的ADAR编辑位点位于非编码区域,位于内含子区域的位点代表了已注释和新发现位点总数的大部分(图 5b,c)。vlincRNA中的位点则代表了相对未开发的新位点(已注释3.7% vs. 新发现25.5%,图 5b,c)。总而言之,研究者发现内含子和vlincRNA对真正的编辑位点有强烈的偏好,基于已注释位点的odds ratio分别为4.7和4.2(图 5d)。然而,编辑位点仅在内含子、vlincRNA、3' UTR中显著富集,在5' UTR中也有少量富集,在其他非编码区未显示富集(图 5d)。此外,
虽然内含子转录本和vlincRNAs似乎包含新发现的位点,但大多数3' UTR 或5' UTR中的ADAR编辑位点可能已经被注释了。
通过对阳性和假阳性RNA编辑位点上下游5bp的motif分析发现Sanger验证阳性与阴性位点相比,编辑位点5' 毗邻的位置上有明显的尿嘧啶(U)偏好。而该流程预测的所有位点的该位置同样明显富集U,与Sanger阳性位点和此前的研究结果一致,进一步支持了该流程最终预测位点列表的可靠性(图 5e,f)。
图5. 癌细胞系中A-to-I编辑位点的基因组分布图谱
(图源:Wang,
et al.
,
BMC biol
, 2023)
尽管本研究提出了较高置信度的已注释RNA编辑位点列表,但仍有25.5%的位点真实性存疑。将仅在一个样本中检测到已注释位点同时纳入考虑,那么K562细胞系中可能验证失败的已注释位点的总比例至少为48.7% (图 5)。这些潜在的假阳性位点可能是由PCR扩增、cDNA合成或NGS等引起的错误或代表了除K562以外的一些样本中的真实ADAR位点,同时仍然代表K562中的错误位点。为了解释这种可能性,作者使用了来自Cancer Cell Line Encyclopedia (CCLE)的K562 RNA-seq数据集,并使用与CLAIRE发表的相同的分析程序对CCLE K562样本进行编辑位点预测,有很大比例(38.0%)在130个K562样品中无法通过三种方法中的任何一种检测到(图 6),
这一比例与上述已注释位点中可能的假阳性位点比例(48.7%)一致。这些结果表明,公共数据库中确实存在很大一部分有问题的ADAR编辑位点。
图6. 公共数据库中非重复序列A-to-I编辑位点的最小假阳性比例估算
(图源:Wang,
et al.
,
BMC biol
, 2023)
文章结论与讨论,启发与展望
综上所述,本研究提供了一个可用于生成哺乳动物细胞中真实RNA编辑位点的流程及一个全面且经广泛验证的人类癌细胞系中A-to-I编辑位点的列表,并分析了真实的RNA编辑位点的分布。由于绝大部分位于蛋白质编码外显子区域的候选位点都是假阳性位点,关于RNA编辑的结论,尤其是基于位于蛋白质编码区域的编辑位点的结论必须非常谨慎地解释,并且需要Sanger验证结果的支持,而大部分经过验证的真实的RNA编辑位点位于非编码区域。由于大部分非编码转录本的功能和生物相关性都是未知的,相应的在这些转录本上的大部分编辑位点的生物学重要性也是不清楚的,但我们相信正在进行中的在“RNA暗物质”中发现新的编辑位点至少具有两方面的重要作用:第一,真实的、已验证的ADAR编辑位点的集合对于训练新的更有效的算法是极具价值的,尤其是对于依赖于深度学习算法的技术。其次,与A病相关的编辑位点,即使是那些功能尚不清楚的编辑位点,也可以代表潜在生物标记物的来源。
本方法是基于大量Sanger实验验证,但是目前数据是基于人类癌细胞系K562,在其他物种中是否一致仍然有待继续研究。本研究表明从NGS数据中检测真正的RNA编辑位点是一项很复杂的工作,即使是已发表在RNA编辑位点数据库中的已注释位点的假阳性率也很高,新发现的编辑位点更甚,它依赖于使用的分析方法和位点所处基因组位置。值得注意的是,本研究的现有分析平台,主要依赖于三种已发布的RNA编辑位点的预测方法,但是对不同基因组特征的准确性的推断对其他预测方法是否一致需要进一步的研究。开发新的机器学习的方法来提高预测的准确率,这将是未来需要努力的重点之一。
上一篇:
证实了液氮冷冻可以高效的保存大型哺乳动物的心肌结构和功能
下一篇:
揭示组蛋白去乙酰化酶HDAC6参与DNA双链断裂修复新机制
已经到最底了
技术支持:
库价化学
Copyright © 2024北京螽斯羽生物有限公司 备案号:
京ICP备2023018288号-1