
白内障是全球首要致盲眼疾,其形成的原因来自遗传突变,应激因素作用及衰老多个方面,分子机制十分复杂。但重要的分子特征是上述几个方面因素造成了蛋白质损伤,而损伤的蛋白变性堆积导致白内障发生。之前的研究表明,在晶状体中存在的α晶体蛋白能够发挥类分子伴侣的作用[1],在蛋白受损的情况下,其能够与受损的蛋白质暂时的结合,维持其溶解度,但是α晶体蛋白无法同真正的分子伴侣一样,通过水解ATP来修复受损蛋白,随着细胞中受损蛋白的积累,越来越多的α晶体蛋白被结合而沉淀,最终导致晶状体透明度降低及白内障的发生[2]。因此,晶状体保持透明需要高效且有修复功能的真正分子伴侣蛋白。
利用中山眼科中心研究平台WES 微量分析技术,首先研究者对正常透明晶状体和一百多例不同年龄段白内障病人晶体上皮细胞中多种HSP90亚型(HSP90a, HSP90β, TRAP-1 和GRP94)和不同HSP(HSP90, HSP70, HSP60和HSP40)表达水平进行定量分析,结果表明HSP90β是其中表达丰度最高的热休克蛋白,且在白内障病人中其表达显著下调。其次研究者利用同样的技术分析了不同年龄段小鼠晶体上皮中上述蛋白的表达模式,结果表明HSP90β是表达最强烈的热休克蛋白且随着年龄的增长其表达显著下调。
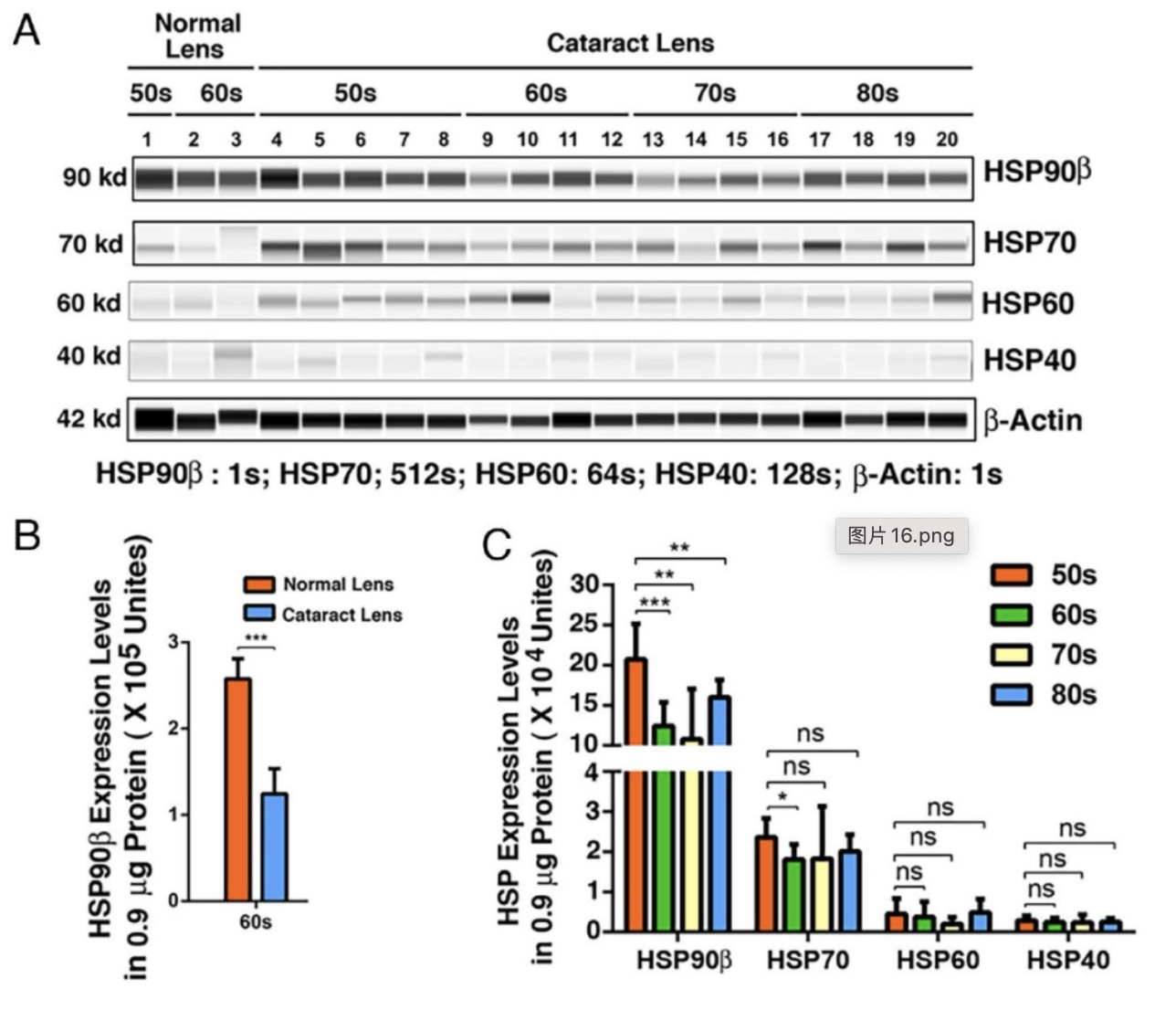
图1 HSP90β是晶状体中的关键分子伴侣
(图源:Fu.et al., PNAS, 2023)
随后,本文对HSP90β在晶体上皮细胞中高表达以及在年龄增加和白内障发生的过程中表达下降的原因进行了探讨,结果表明HSP90β的启动子上存在多个Sp1家簇转录因子结合位点。 利用多种分子生物学技术,作者阐明HSP90β在正常晶状体中的高表达主要是受到Sp1家簇转录因子Sp1和Sp4的正向调控。 HSP90β而非其它HSP90亚型敲低导致白内障和小眼症,这一缺陷可以由HSP90β基因回补更正,但不能被其它HSP90亚型的回补,同时在Sp1及Sp4敲低的斑马鱼中也出现了同样的表型。说明HSP90β是调控晶状体发育的关键基因,且其表达受Sp1及Sp4调控。
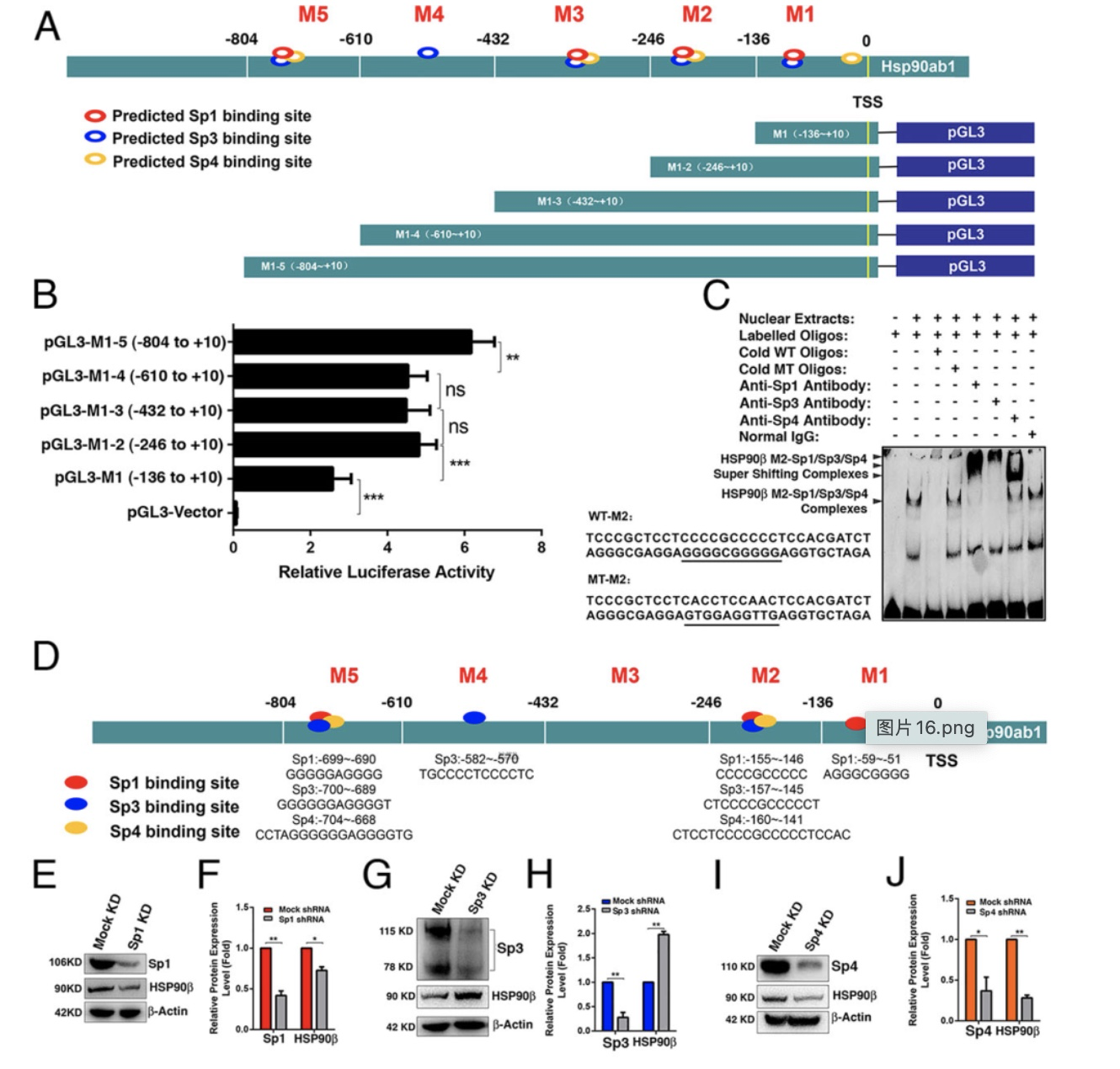
图2 Sp1家族转录因子调控HSP90β的表达模式
(图源:Fu.et al., PNAS, 2023)
为了深入探究HSP90β调控白内障发生的分子机制,研究者通过质谱分析识别出了一个之前未被报道过的HSP90β的靶蛋白——CHMP4B。通过斑马鱼模型,研究者发现HSP90β或Sp1/Sp4沉默导致CHMP4B上调,因此增加有丝分裂指数,而分裂的细胞因凋亡程序的激活,导致发育中的眼组织(包括眼晶状体上皮细胞)大量凋亡。

图3 HSP90β通过与客户蛋白CHMP4B调控晶体上皮细胞周期
(图源:Fu.et al., PNAS, 2023)
RNAseq分析及随后的QRT-PCR和蛋白质印迹实验证实HSP90β通过与客户蛋白p53相互作用抑制p53介导的凋亡通路。敲低HSP90β导致p53-Bak/Bim凋亡途径激活,从而使分裂的晶体上皮细胞走向凋亡。
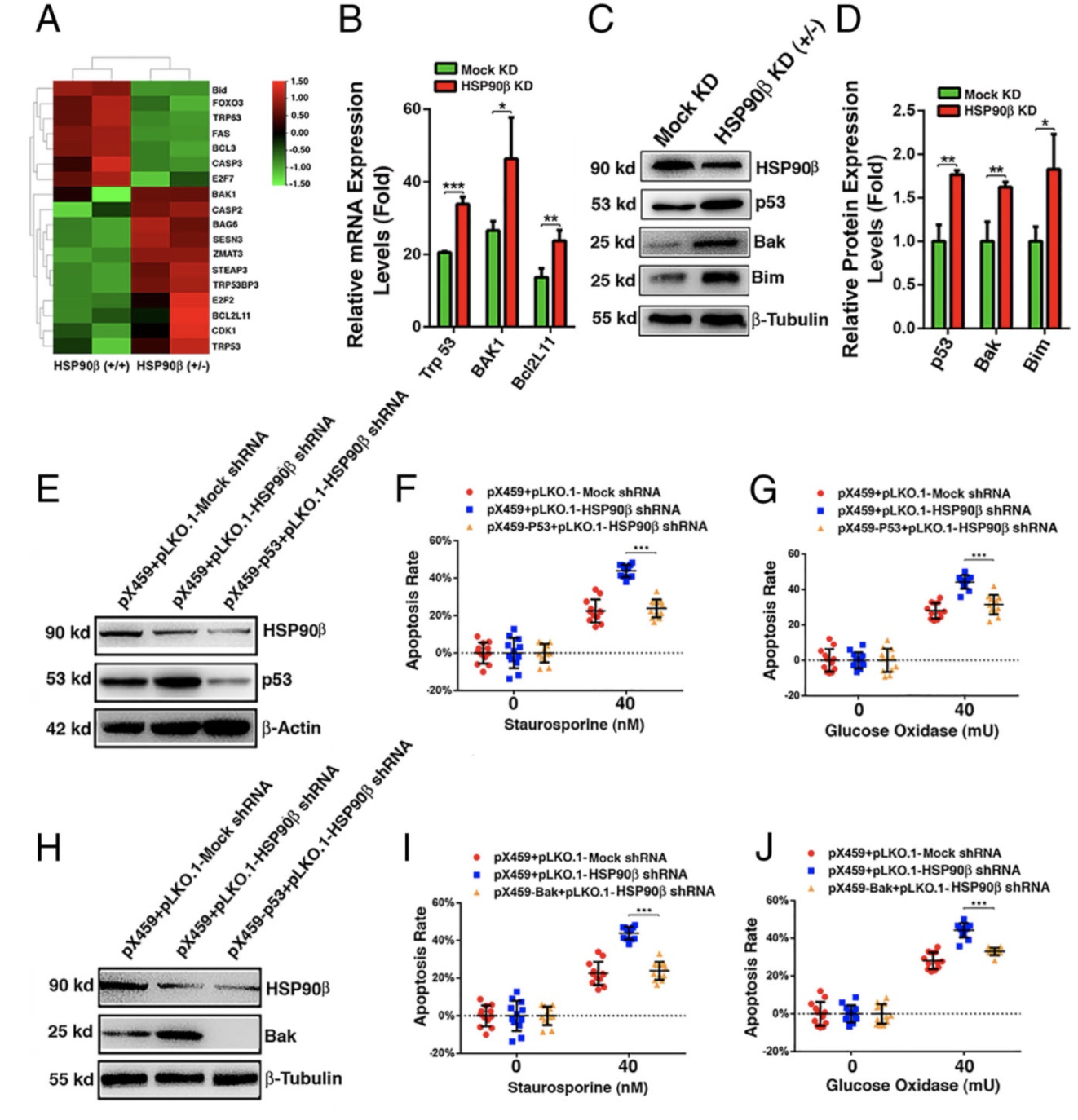
图4 HSP90β通过客户蛋白p53控晶体上皮细胞凋亡
(图源:Fu.et al., PNAS, 2023)
此外,对于热休克蛋白的关注往往集中在癌症、视网膜疾病及神经退行性疾病中的功能,本研究阐明了HSP90β在晶状体发育及白内障发生过程中的功能机制,为白内障的发病机理和临床治疗提供了新的研究方向。
